pFind团队的追求与道路
2012年, Nature Methods发表中科院计算所pFind团队与北京生命科学研究所合作完成的交联肽段质谱鉴定软件pLink,这是中国团队的蛋白质组学研究成果首次突破 Nature子刊。之后的几年中,中国蛋白质组学研究成果不断在Nature子刊上发表。其中,pFind团队与复旦大学合作完成的完整糖肽质谱鉴定软件pGlyco 2、独立完成的常规肽段质谱鉴定软件pFind 3分别于2017、 2018年发表在Nature Communications、Nature Biotechnology。2019年7月,新版交联肽段质谱鉴定软件pLink 2在Nature Communications发表;10月,pFind团队领衔完成的《蛋白质组海量质谱数据深度解析关键技术及应用》成果荣获2019年中国计算机学会技术发明一等奖。pFind团队师生总结回顾其研究历程,与大家分享。
1. 贺思敏:pFind 团队的追求与道路(2017年2月19日)
2. 曾文锋:pGlyco 2 的研究历程回顾(2017年10月13日)
3. 迟 浩:pFind 3的研究历程回顾(2018年12月3日)
4.
陈镇霖:pLink 2
的研究历程回顾(2019年9月30日)
发件人: He, Si-Min [mailto:smhe@ict.ac.cn]
发送时间: 2017年2月19日 23:54
收件人: allpfinders@ict.ac.cn
主题: 转发: pFind 3软件在北京生命科学研究所董梦秋实验室的应用报告
诸位毕业和在读的pFinder,
转发董老师为迟浩863课题结题所写的pFind 3用户报告,请看附件。读了报告,我浮想联翩。
——学习的目的是什么?学术的目的是什么?
以我为例,从1975年小学一年级到1997年博士毕业,学习了20+年,为了什么?一个博士学位?一份工作?从1997年博士毕业工作到现在,算是在学术道路上走了20+年,又为了什么?一个研究员头衔?一个 PI位置?一篇顶刊文章?
我喜欢“学以致用”这简朴的四个字。什么“用”?自己用还是他人用?引用还是应用?显然是后者。你猜假设pFind 3在Nature Methods发表文章我更高兴,还是pFind 3软件在董老师那里用起来我更高兴?虽然前者看起来高大上、更有显示度,但是我选择后者,因为前者只是显示pFind自身有力量,后者则显示pFind对他人有用。
——为什么要做软件?为什么不能只做算法、只写文章?
人的精力有限,写软件多,写文章就少。pLink第一博士小樊毕业时我请他临别建言,他特别提到咱们组写软件、推广和维护软件耗费不少精力,影响写文章、影响毕业。我相信小樊的意见在咱们组内具有一定普遍性。实话说, pFind组文章真不算多,而且学生毕业、员工升职,文章都是硬指标、第一指标,我也不能说从来不为此着急。
不过思来想去,我还是觉得软件不能丢。
想想咱们组的明星软件pLink,如果停留在2012年Nature Methods发表时的算法Demo水平,没有小樊、佳明、吉澧持续的维护、更新,那么我们今日就看不到国内外同行独立应用pLink软件发表 46篇文章,其中IF达到PNAS以上的18篇,Cell/Nature/Science正刊文章7篇。同样,迟浩为主开发的pFind 3,如果仅仅满足以 Demo软件承载核心算法,即使也可能实现冲顶,但是 Demo软件不可能在董老师那里真正用起来,董老师一定会怀念其旧爱ProLuCID。
这些年我身处交叉领域,偶尔也回味、审视相对单纯的计算机领域的生活。大家知道,我其实是被动走上pFind之路的,在此之前有7年时间研究交换机调度算法,也算合作发表过一篇网络领域顶刊ToN文章。我曾经想过设法使这篇文章的成果进入到网络产品,比如华为、思科的交换机中,但是显然,这太难了。我当时的合作者说,发表文章就可以了,想应用那得等十年,这是规律。可是我哪等得了那么久?这些年所里同行投稿顶级会议也不断有突破,被多少多少同行下载、引用,其实我想也有类似的问题,那就是引用再多不一定是应用,也很难立即应用看到结果。
但是pFind研究很不同,我们不仅可以发表高水平文章,而且可以把研究成果以软件的形式直接推广到最终用户,比如董老师这里,比如发表CNS文章的同行那里,看到开花结果。我喜欢这种立竿见影的效果!咱们很多同学毕业之后在各大公司也做软件,比如佳明在亚麻、罗兰在微软、吉澧在百度,但是这些公司做的软件太大了,个人的力量占比太小,比如吉澧可以说“ pLink 2.1是我做的”,但是不好说“Baidu 2.1是我做的”,这是在pFind这个小团队、“小公司”做软件的另一个优势。
假期阅读看到一句话,美国计算机天才克雷说:“可以造出一个速度快的CPU,却很难造出一个速度快的系统。” 只有 CPU是没法用的。
——为什么做软件的同时,一定要坚持做算法?
因为没有CPU也是不行的。
在交叉领域做研究,特别要警惕丧失自我。当前分子生物学进入组学时代,测序仪、质谱仪分别把基因组、蛋白组数字化,快速生成海量数据,对于数据处理产生巨大的需求,生物学家不得不求助于搞数学、搞计算的人,搞得后者特别容易飘飘然。尤其是生物学期刊影响因子普遍远高于数理科学,学科交叉又属于“政治正确”,所以未来搞生物信息学的人会越来越多。可是这些年我们看过的很多质谱鉴定软件文章,其中只有简单的加减乘除,没有深刻的计算技术。我也听过一些原本受过很好数学训练的人所做的生物信息学报告,很简单的计算和软件就获得生物学家的广泛应用,自以为四两拨千斤,我想报告人已经忘记了自己从哪里来、要到哪里去了。
2016年中国计算机大会有一个特邀报告,瑞士洛桑联邦理工学院院长介绍《如何打造一个顶尖计算机学院》,提到“四个谨慎”,其中第四个是:“4. 谨慎对待跨学科合作:如果合作是平等的,那跨学科合作是好事情。而‘不好’的合作经常出现,例如有时候生物学者想要找一个懂计算机的助手,于是就来计算机系找人来帮忙。计算机本身是个独立的学科,学者不是为了成为他人的附属帮手而存在。我们在招聘时需要员工具备本专业应有的专业素质。”
其实生物学家想找个附属帮手是完全正当、非常可以理解的,而且能做好这个帮手也远非想象那么容易,比如pFind从2003年立组之初到2015年持续奋斗13年才赢得了进驻董老师实验室的机会。但是如果仅仅满足于维护pFind 2,而不是从内核到界面脱胎换骨,那么pFind根本无法替代ProLuCID。坦率地讲, pFind 3的研制动力,首先来源于迟浩为首的pFinder的梦想,来源于他们对pFind 2的不满足、不满意。在董老师用户报告中,大家只能看到软件的外在性能,却看不到pFind 3内部算法的风采。我举一个例子,报告中提到pFind 3结果展示特点时说:“样品总体上特异性酶切比例、哪些修饰出现最为频繁…一目了然。”这两个功能等效于pFind 2软件同时指定非特异酶切、指定单个位点上允许设定Unimod上千种可变修饰,搜索空间比常规大10万倍,pFind 2根本跑不出结果,即使跑出来结果,精度也会一塌糊涂;而 pFind 3则可以在pFind 2常规搜索的时间内跑完,相当于内在提速10万倍,而且保持并提升了精度!我再举一个例子:当年阅读计算蛋白质组学领袖Pevzner团队的文章,看到他们利用AC算法实现肽到蛋白归并,我为自己的无知而难过, pFind 2当时马上借鉴了 AC算法;但是今日迟浩的pFind 3已经完全抛弃了AC算法,设计了更巧妙、更高效的算法。这些算法,体现了我们计算所人的专业素质,这也是我们pFind团队的核心竞争力!
我们组这么多年来,一直比较强调算法的重要性,追求软件的算法特色,坚信算法是有力量的。但是这个追求过程很痛苦,因为应用问题的算法核心在哪里,往往隐藏得很深,pFinder常感到有劲儿使不上,而硕士时间很短,即使博士时间也并不够长。比如博士生李德泉开创的索引软件 pIndex,经过接班人李由的硕士论文研究发展,才终于变成组内的公共技术;经过迟浩的硕博连读创新再造,才成为pFind系列软件的第一核心技术。再比如袁作飞硕博连读开创的预处理软件pParse,经过接班人邬龙的硕士论文创新再造,才终于有了清晰的计算模样,现在成为pFind系列软件的必备前端。
虽然我从博士研究开始,专业方向就是“计算机应用”,但是只有pFind的十多年我才真正理解了、真正做到了“计算机应用”。与pFind之前十多年相比,最鲜明的特色就是:pFind的所有研究问题都来源于合作伙伴一线科研需求,而不是别人的Paper。为此我们花费了大量时间、甚至是最宝贵的年华,从原始问题中提炼计算模型,虽然似乎始终不如大数据、深度学习、云计算那么高大上,而且我也的确放弃了从交叉科学中发展计算机科学、发明新数学的念头(不是不可能,而是我没有那个大智慧),但是我始终认为:把理论方法应用到实践绝非轻而易举;学不易,行亦难,甚至行益难。打个比方,学习《算法导论》不容易,那么应用《算法导论》容易吗?我现在觉得后者更难,因为能从书本上学到的东西,都不算难。
——pFind 3的下一步?
关于pFind 3,我一直记得刘超2014年的一句话:“我一直想开发像现在pFind 3.0这样的软件。” pFind 3进驻董老师实验室,是pFind新时代的开始:在 pFind 3的激发下,今年我们还将发布交联鉴定新版软件pLink 2和完整糖肽鉴定新版软件pGlyco 2,预期会给领域带来小小的惊喜甚至震动。我最看重的是,这一代软件已经具有更为清晰的计算模型,意味着计算技术可以发挥更大的作用。迟浩正在赶写pFind 3文章,期待他的喜讯,但是也坦然接受噩耗。无论年终是喜讯还是噩耗,生活仍要继续,那么朝着什么方向呢?
回想克雷的名言,目前pFind 3其实已经不是一个软件CPU了,而是一个软件系统。除了最核心的新版搜索引擎pFind,还有新版后处理软件pBuild、pLabel,新版前处理软件 pParse,新版集群软件,特别是新增定量软件pQuant,实现了pFind 3定性定量一体化。不过,相比董老师实验室完整的质谱鉴定干湿流程而言,pFind 3仍然只是一个CPU,而这个CPU想充分发挥作用,还必须对整个流程系统全面考察。
年前我带刘超去拜访董老师,发现pFind 3想一展雄风并非那么简单。比如董老师原先的软件系统不仅有Yates的ProLuCID,还安装有另外一方的修饰位点定位开源软件。此外,由于最基础的质谱采集DDA流程的固有缺陷, MS2谱图采集不充分,多轮数据分析下来有效数据很少,无法获得可信的生物学结论,因此董老师很希望借助Mann团队MaxQuant软件的MatchBetweenRun功能来弥补缺陷,但刘超分析后发现这不太好马上支持,最好从流程设计之初就考虑这些问题。
所以,不要以为pFind 3在董老师那里稳定运行一年就万事大吉了,即使pFind 3是流程的长板,流程的性能还是由其最短板决定的。所以,要从全流程的角度来重新考察pFind软件设计,强调干湿融合。就好比设计计算机,体系结构是硬件,操作系统是软件,必须协同设计、相互补台。为了最终发挥 pFind软件的价值,我们必须解决最后一公里的问题,或者先跟踪Mann的流程技术,或者直接发展新技术比如DIA,这都需要我们跳出 pFind软件当前的舒适区,去学习、理解前端实验流程,必要时做干湿流程再造,这就是pFind 4的蓝图。
希望毕业的pFinder,能从这份报告中感到你们当年开创pFind软件的努力没有白费。
思敏
2017年2月19日

最“甜蜜”的小皮——pGlyco的研究历程
曾文锋
pGlyco是pFind软件家族的新成员。pFind软件曾经在中科院动物所质谱室李晓明老师那里服务多年,深受李老师喜欢,他给 pFind起了一个中文名字:小皮。pGlyco鉴定对象是糖肽,比pFind鉴定的肽多了一个糖,听起来很甜蜜,可做起来的感觉却是…
〇、才入虎穴,又进狼房
pGlyco对我来说,完全是个意外。2013年年中,贺思敏老师突然找我面谈,说让我和硕士生吴建强一起去复旦杨芃原老师实验室呆一个月,协助复旦完成pGRIP的项目(pFind+复旦张扬博士开发的GRIP)。实际上之前我们早就确定了去复旦的人选,那就是建强。因为早前pLink的合作也是由二年级的硕士生开辟的,所以对于糖肽,我觉得应该没我什么事。
那时,我的pDeep研究才刚刚开始。恰逢DIA(一种新的质谱数据采集方式)的热潮也刚刚被SWATH掀起,我也才刚刚完成了DIA综述,打算进入DIA这个热门的领域。更重要的是当时我对糖肽一窍不通,所以对我而言,糖肽是一个异常困难且不熟悉的问题。然而去一个月就回来了,只是去协助建强开发,也不算换方向。而且组里其他博士师兄们都忙着自己的课题或毕业论文,就我最闲了,所以还是去吧。
没想到这一去,就是一年,而这糖肽一做,就是数载。
一、巧妇无米,好友借光
我们拖着行李,来到了复旦,杨老师给我们安排的是一间老师办公室,我们觉得和老师们一起办公不太方便,就搬到了复旦IBS大楼的地下室。当时张扬博士和湖南大学的谷惠文博士,是我和建强在地下室的两位难兄难弟。刚开始的两周,除了张扬博士传授我们GRIP的基本方法之外,其他时间我们都在按部就班地开发pGRIP。pGRIP是切糖链的流程,其中切掉糖链后由pFind鉴定肽段,GRIP算法则通过 pFind鉴定的肽段列表在完整糖肽的CID谱图上搜索并鉴定糖链。如果按照杨老师和贺老师开始的设想,我们其实能够在不到一个月的时间内完成pGRIP。
然而还没有完成pGRIP,贺老师就发来短信,大意是说能不能开发一个不切糖链的糖肽鉴定流程。这个要求,对我这个当时五糖核心都理解错了的门外汉而言(我当时以为五糖核心就是Hex、HexNAc, NeuAc,Fuc和Xyl这五种基本糖单元),太困难了。另外在算法开发上,我们手里也并没有不切糖链而且能够同时鉴定糖链和肽段的数据。对计算而言,数据就是米,是一切的基础。
后来看了几篇文章,发现了HCD-pd-ETD这项技术。终于通过贺老师的穿针引线,我们联系到了当时还在Thermo公司的贾伟博士和张伟博士,他俩为我们提供了第一个不切糖链的HCD-pd-ETD的数据。
即使有了数据,软件的设计依然是最困难的环节。由于pGRIP算法无法处理不切糖链的数据,所以针对HCD-pd-ETD谱图,必须换一种方法;我们思来想去,最终开放式搜索方法成为了我们的首选。为了设计pGlyco的开放式算法,我和建强有段时间在地下室不停地画着各种流程。低能量HCD谱图中,一般糖肽的Y离子(糖碎片+肽段 )比较丰富,但是ETD的肽段碎片信号却有好有坏,难以估摸。所以,我们觉得应该先开放式地搜糖链,后通过母离子与糖链的质量差,搜索并鉴定肽段。建强完成糖链搜索部分,我则完成肽段搜索部分,就此完成了pGlyco 2的雏形。(由于基本没有较大规模的基准数据,所以这里只有雏形,打分就是数数匹配谱峰数,FDR即错误率也没得入手估计。可能跟其他软件的开发不同,我们的 pGlyco 2的软件框架比pGlyco 1更早诞生。)可惜的是在当时HCD-pd-ETD数据上鉴定结果太少太少。
虽然算法完成了,但是我们却没有想出更好的质谱实验方案。贺老师认为我们应该继续深入这方面的研究,本来说好在复旦只呆的一个月,变成了无限期。对我们做谱图搜索的人来说,没有数据,前面的道路完全是黑暗的。
二、明灯指路,窃玉何妨
几个月后,杨老师博士生刘铭琪师兄刚刚休完陪产假,回到了实验室。建强,刘铭琪和我三人一起,拿着pGlyco 2这个锤子,到处找钉子。比如HCD和CID一母双谱,开放式检索CID谱图,在HCD谱图中找Y1离子确定糖的身份;比如用CID鉴定糖链后,用 Targeted(靶向)的方式碎裂Y1离子的三级谱等等。这些实验要不就是无法解决肽段鉴定问题,要不就是在老仪器上实验效果很差,最后都没有什么结果。2013年年终,贺老师对我们三人的成果非常不满。在当时的p系列软件中,pFind 3已经支持了意外修饰和非特异酶切的快速开放式搜索, pLink-SS正在二次冲击Nature Methods,pTop也在罗兰师妹手里由0变1。但是pGlyco这边,我们连长得像样的不切糖链的RAW( Thermo质谱仪原始数据格式)文件都没能呈现给大家。
虽然贺老师在学术上对我们要求极其严苛,但是在其他方面他总是不遗余力。我记得在复旦的第三个月前后,我和建强的老笔记本实在是运行不顺畅了,就跟贺老师说了此事。没过多久,贺老师和迟浩师兄就一人背着一台新买的笔记本,第一时间交给了我们。他们当时应该也背着自己的笔记本,所以一人是背着两台重重的笔记本,从京城来到魔都。虽然贺老师这次来魔都,在交流会议上对我和建强的工作进行了批评教育,但是估计他也知道,巧妇难为无米之炊,所以在很长的一段时间之内,他也一直在操心着“米”的事情。这一点从贺老师搜寻HCD-pd-ETD数据上就可以看出来。有几篇糖肽相关的文章,比如台湾邱继辉教授的Sweet-Heart系列文章,都是贺老师第一时间搜到并且转发给我们的。
在我们三人加上贺老师的不断推进下,pGlyco湿实验方面的研究,终于迎来了转机。
pGlyco的第一个转机,就是Fusion新仪器。刘铭琪师兄用Fusion测试Targeted三级谱时,脑洞稍开,发现 Fusion下可以直接用DDA的方式碎裂三级。拿到DDA-MS3的数据,我们基于pGlyco 2的模型开发了pGlyco 1。当时,大家本来想把宝压在pGlyco 1身上,但是三级谱的速度和灵敏度的问题,一直不容易解决,所以三级谱即使有 DDA,也不能说是一个优雅的方案。因此对于pGlyco 1,我们没有再去测试复杂样品,直接使用简单样品写成了一篇小文章。
pGlyco的第二个契机,就是军科钱小红老师/应万涛老师课题组发表的用阶梯能量碎裂切糖链后的核心岩藻糖化肽段的方法。贺老师仔细阅读过Mann的AIF那篇文章,阶梯能量对他来说不是什么新概念。我也写过DIA综述,阶梯能量对我也不是什么新方法。我们知道HCD既可以碎裂糖,又可以碎裂肽,但是我们并不知道HCD能将糖碎裂到什么程度,将肽碎裂到什么程度,可能领域给我们的第一印象,就是 HCD不能同时碎裂糖肽。而钱老师和应老师的这篇文章,激起了我们心中的波澜,为我们指明了方向。后来我们找到了一些低能量HCD(25能量)的谱图和高能量HCD(40能量)的谱图,我专门开发了糖肽标图工具gLabel来看看这些糖肽匹配,发现低能量上能够看到较为丰富的糖链碎片,高能量上则可以看到不少肽段的碎片离子。就是它了!!那就是我们那时候的主要感觉。
这里我还想再提一下gLabel,我个人特别喜欢这个工具,我的博士论文、pGlyco 2的文章中满是gLabel画的谱图,然而它却是最不容易写成文章的工具。外围工具和引擎核心并驾齐驱,是pFind组里的传统,比如 pFind搜索之前,用pParse校准母离子,搜索之后用pLabel查看匹配情况。pLink搜索前,也用的pParse,之后也用的pLabel(拓展版)。Top-down的 pTop也是如此,拓展版的pParse和拓展版的pLabel。如果没有谱图标注工具,搜索引擎利用打分很容易骗过我们,我们开发人员就很难对算法进行进一步改进。所以谱图标注,是pGlyco不可或缺的关键环节。有了pLabel作为基础,开发一个糖肽的标注工具 gLabel,开始是很简单的,当然后来还是很不容易的。糖肽标图和肽段标图是两个截然不同的形式,主要问题是糖链是天线结构,其碎片不能简单用y1、y2、y3来表示,所以对糖链的标注需要占用较大的绘图空间。这部分我和刘铭琪师兄一直在考虑标注文本的放置,现在其实也没有做得很好,我们一直在改进。但即使是简单版本的 gLabel,已经足够我们看出不同能量的HCD是可以出现大量糖碎片和肽碎片的。
那时我已经在复旦呆了将近一年,刚回计算所,刘铭琪师兄也主动跟我们一起来到了北京。贺老师、刘铭琪师兄和我,在贺老师办公室讨论了许久,决定来一次地毯式能量搜索,看看糖肽在什么能量下出糖碎片,什么能量下出肽碎片,怎么组合能量比较合理。这个方案杨老师在行动上很支持,但是一开始他还存在一些质疑,他觉得刘铭琪师兄和我这是在做蒙特卡洛(可能是指用不同能量随机将糖肽打碎吧),实际上我们搞计算的,在问题原理明确之前,很喜欢蒙特卡洛。我们后面做了地毯式的能量实验,写了一些脚本专门分析地毯式能量,同时分析不同单能量的三三组合(因为质谱仪只允许同时设定三种碎裂能量)。分析完谱图信号特征后,我们得到了20-30-40为最优阶梯能量组合,在此阶梯能量下,糖和肽的总体信号最好(注意,不同的仪器可能不一样)。如果可能,地毯式能量 HCD碎裂在一张谱图里面肯定最好。做了一些标准品的阶梯能量数据后,结合gLabel谱图标注和手工验证,我和刘铭琪师兄一起对pGlyco 2进行了疯狂的调优,比如如何通过作为糖链特征的氧鎓离子确定糖肽谱图、如何通过氧鎓离子判断NeuAc的存在、如何支持含有NeuGc的样品等等。由于当时的数据一直在换,所以后来干脆用机器学习的方法来做打分参数的自动优化,包括如何利用 Y1离子的信号、如何利用五糖核心离子的信号等等。
三、万事俱备,以湿见干
有了阶梯能量和pGlyco 2的算法,怎么通过湿实验的设计,来证明我们干实验(也就是算法)的优势呢?刘铭琪师兄就开始考虑生物故事方面的问题。他认为,当时的糖肽鉴定的主要问题是通量问题,所以我们要做出一定数据量,使整体的通量上一个台阶,为领域创造一个纪录。如何选择生物应用方向,非我强项,所以我认为算法上还需要更进一步,比如糖肽的FDR估计问题。那时刘铭琪师兄刚刚写完一版pGlyco 1的文章,所以我在优化pGlyco 2算法的同时,也接过 pGlyco 1的文章写作任务,一方面让刘铭琪师兄和杨老师博士生方盼专心做高通量的质谱实验,另一方面也是计算所毕业要求有两篇SCI一作文章,而我已经是博士第五年了。要不是毕业时间比较紧张,我觉得我们会将pGlyco 1投在JPR这类期刊上。
刘铭琪师兄最后决定用老鼠的各个脏器作为基本样品,不分馏,重复几次质谱实验,看看我们能冲刺到多少糖肽(考虑过血清,但是血清的问题是糖蛋白丰度差异太大)。当时我们用的是上海蛋白质研究中心黄超兰研究员实验室的Fusion。不知为何,那台仪器无法做阶梯能量的HCD实验(后来知道是质谱仪的bug),所以我们只好以HCD20+HCD40的一母双谱实验代替阶梯能量。实验过程中刘铭琪师兄还发现了提高上样量等方案能够提高肽段离子的信号强度等一系列质谱参数优化方案。最后,我们终于在量上突破了5000条非冗余糖肽的大关!
就在刘铭琪师兄做通量实验的同时,我工作的侧重点还在计算问题上,那就是如何估计糖肽的FDR。
四、糖错肽错,左右犯难
很多糖肽鉴定软件,都只去估计肽段部分的错误率,然后以此做为糖肽的错误率。大家肯定知道这么做是不完全正确的,但是一时之间也想不出更好的方法,包括我们pGlyco。当时面临的主要问题是,如何估计糖链部分的错误率?而在估计完糖链部分的错误率后,如何估计糖肽整体错误率?
糖肽FDR估计问题,其实贺老师在刚让我做糖、我可能还没去复旦的时候,就已经向我提出来了。当时我给组里写过肽段鉴定评价算法的综述,其中主要部分就是在讨论如何估计肽段的FDR,所以我做糖肽FDR的估计,也算义不容辞。可以这么说,错误率估计问题,一直是蛋白质组学领域内的最关键但是又很难突破的计算问题之一。 pLink文章解决的一个关键的计算问题,其实也就是交联肽段FDR如何估计。pGlyco无法像pLink那样可以直接基于TDA方法(Target-Decoy Approach,目标诱饵库方法)巧妙地推导出公式,因为基于蛋白 Reverse(序列反转)的Decoy(诱饵)策略只适用于肽而不适用于糖。糖是树形结构,它的Reverse是什么?期间建强和我也都想了一些糖链结构Decoy的方案,但是都被我们否定掉了。结构不能Decoy,那么谱图能否Decoy?第一个想法是,我们将实验谱图进行 Decoy也许就可以了。但是这个想法逻辑上说不通,因为一旦谱图被Decoy了,那么肽段的匹配也就变成了错误匹配,此时我们得到的是二者同时错误的匹配,而不是单独糖链错误的匹配,这个问题,真是左右为难!
这个问题的解决,我也忘记了是什么时候的事情了。反正贺老师之前让我写的肽段鉴定评价算法的综述,在最后终于派上了用场。传统的TDA不行,就参考Decoy实验谱的方法;Decoy实验谱的想法行不通,就将TDA和Decoy实验谱的想法结合。我们用TDA的最终目的是什么呢?其实就是想获得Decoy的匹配打分,以此估计 Target部分的FDR。基于Reverse的Decoy,它首先生成了一条反转的肽段序列,然后生成反转序列的理论谱图,再与实验谱图进行匹配获得Decoy打分。Decoy实验谱的方法就是将实验谱图进行变换,然后再与 Target序列进行匹配获得Decoy打分。那如果我们不在序列上进行Decoy,而在理论谱图上进行Decoy,那我们岂不是可以在不改变实验谱图的情况下也能获得Decoy匹配?这个想法的基础是,直接通过Target生成理论谱图,然后 Decoy这个理论谱图,再与二级谱进行匹配获得Decoy打分。只要Decoy策略设计合理,这个想法可以同时适用于线性的肽段、树形的糖和首尾相连的环肽。然而此时Decoy与Target的随机匹配我不敢保证一定是1:1,所以引入了有限混合模型 FMM算法进行比例的校正。我在肽段鉴定上尝试了这个算法,发现效果跟传统的TDA是完全可比的;然后再在标准样品上测试了糖肽的数据,与实际错误率的偏差也不大。至此,pGlyco终于解决了糖链部分FDR的估计问题。
即使糖的FDR和肽的FDR可以分别估计了,那糖肽的FDR怎么估计呢?两个加起来?加起来合理吗?有数学解释吗?这是我脑海里挥之不去的问题。“肽对并不意味着糖对、肽错也不能说明糖错、但是肽错的情况下糖错的概率会比较高 ……” 左边的肽和右边的糖,这一系列数学、非数学的关系,犹如相互缠绕的线,在我的脑海转来转去。糖肽FDR的问题,至少困扰了我几个月的时间。
不知道哪天,灵光一现,突然就想通了糖肽FDR的贝叶斯解释,也就是文章中糖肽FDR的数学公式。寥寥几行公式,就能解释糖与肽之间对对错错的数学关系,当时很兴奋,同时也在暗笑自己,这么简单的问题,怎么会想了那么久。有时候问题的答案往往比较简单,只是我自己想复杂了。后面糖肽FDR公式的推导,也为组内复杂鉴定的评价工作,尤其是交联鉴定的评价工作,提供了新的范式。
想到这个公式时,pGlyco 2的NM(Nature Methods)审稿已经进入了第一轮修回。
五、东隅既逝,桑榆非晚
pGlyco 2的NM送审比当年pLink顺利太多,期刊编辑很喜欢我们的工作,不过她先要我们加上与Byonic软件的比较,然后很快就送审了我们的稿件。在我看来,审稿意见不能说太坏,特别特别是编辑的意见(审稿意见我贴在了本文末尾的附录中 )。
我们加入了一些新实验,也加入了新的糖肽FDR公式。可惜由于pGlyco 1文章的投稿没有预先告诉编辑,pGlyco 2后来还是被NM拒稿了(见附录的NM第二轮审稿意见,其中编辑对没有告知她pGlyco 1的工作有些失望)。实际上,pGlyco 1和 pGlyco 2的质谱流程、算法流程等都不相同,所以我们没有在pGlyco 2中提起pGlyco 1,但是总归有一些关联的地方,我们没有处理好。这次投稿过程中,我们对于同时投稿两篇文章没有什么经验,虽然pGlyco 1先于pGlyco 2投稿,但是内容的安排上也许需要更分散一点。总之,此次投稿值得反思。
实际上,即使NM接收,我们几个对当时的数据质量其实也没有那么满意。当时数据是一母双谱而不是阶梯能量,NM投稿文章里面有提,但是估计审稿人不会关注那么细。而且,鉴定量也就5000条非冗余糖肽,没有达到万的量级。
后来杨老师买来了新版的Fusion质谱仪,阶梯能量的bug没有了。刘铭琪师兄和方盼花了数个月的时间重新采集NM投稿所使用的阶梯能量的数据,他们还加入了一些色谱、质谱条件的优化等。新的文章中,复旦曹伟倩师姐也加入了贺老师一直在推动的、从北京生命科学研究所董梦秋老师那边学习的15N和13C标记的实验,来验证我们糖肽FDR估计算法的正确性。另外我们还加入了陷阱库(糖陷阱和肽陷阱),验证了糖FDR控制的必要性。补完实验后,由于我需要准备我的毕业论文等事宜,所以只要不是算法优化上的任务,比如文章写作、后续的一些分析等,都是刘铭琪师兄等人负责的。之后我们尝试了 NBT(Nature Biotechnology)和NCB(Nature Chemical Biology)这两个期刊,都没有送审,返回的意见都是内容不适合在他们期刊发表。最后我们就投稿了NC(Nature Communications)。 NC的第一轮审稿意见出来,审稿人并没有提太多计算上的意见,但是生物方面提了不少问题,其中关键的就是NeuAc和NeuGc的问题(见附录,NC的审稿意见)。审稿意见回来后,复旦方面加入了两个重要分析: 1、 NeuAc和NeuGc在不同小鼠组织中含量不同的验证(主要是切糖链验证);2、组织特异性的糖基化位点图表(刘铭琪师兄的太太设计了文中的五叶草的图)。修回的内容基本打动了两位审稿人,不再有进一步的意见。
贺老师一直推动的利用15N和13C标记来验证鉴定结果可靠性这方面,又是另外一个故事。主要是精准医疗这个概念的兴起,让我们做引擎的对准确性更加关注了。对于一个软件,不是卡了FDR就能说明软件准了,然后基于FDR还不断比较哪个引擎多、哪个引擎少。对于一个软件,首先要验证它的 FDR估计是否准确,因为基于TDA估计的FDR太容易被TDA自身欺骗了,比如不合适地缩小数据库的二次搜索策略等等。最理想的,引擎应该在谱图匹配层次验证每个匹配的正确性,虽然现在的生物和计算技术还没发展到这个阶段,但是15N和13C标记检验技术具有很大潜力。
最后,不管是文章写作、数据质量还是数据分析上,我认为pGlyco 2的NC投稿文章比NM的投稿好太多。刘铭琪师兄也表达过这一想法。贺老师经常告诫,更像是鼓励我们,说“如果当时NM中稿了,对你们不一定是好事,因为pGlyco文章的水平就少了更上一个台阶的机会。”确实,NM投稿时,我们对糖肽 FDR的验证还只能通过人工看谱这种主观的方式。而NC投稿中,我们加入了大量对FDR进行评价的内容,例如15N/13C标记检验和陷阱数据库检验,解释了为什么必须控制糖链FDR的原由,这些内容对糖肽FDR的验证都非常必要,也更加客观。刘铭琪师兄还加入了五叶草等图,在图表设计上也更加用心。因此对于这次NC中稿,我们其实应该更加心安理得一些。最后, pGlyco 2的文章拥有了对糖肽质谱方法和简化实验流程的贡献,对糖肽搜索和FDR估计等算法方面的贡献,和最后怎么客观验证鉴定结果相对准确的评价方法的贡献,所以pGlyco 2值得一篇NC!
六、赠君玫瑰,留手余香
即使文章发表了,pGlyco 2的大石依然在我心里没有落下。主要是因为发表意味着pGlyco 2马上就要面临更加严峻的挑战,那就是实际用户。我们遇到的很多实际情况是,用户使用我们的软件,却达不到我们文章中提到的性能,而更糟糕的情况则是软件跑不通。所以要求软件的参数尽可能少,让用户能够傻瓜式配置。pGlyco 2离傻瓜操作还有很远,一直想写一个C#版的pGlyco界面,但是由于太忙抽不出时间,现在只提供了一个简易版的 Python界面。
如果pGlyco 2能够帮助生物化学家们完成他们的研究,那将是比文章发表更加令人愉快的事情。
2017年10月13日

千淘万漉虽辛苦,吹尽黄沙始到金——pFind 3的研究历程
迟浩
自Open-pFind发表以来,一直记得要写篇总结说一下整个经历,但由于种种原因一直拖到今天。眼看2018就要过去了,我想还是趁着手热,赶紧写了,然后,继续投入到pFind 3+的研究工作中去也。当然,我也借这篇文字解释一下, Open-pFind是一个算法,一个开放式搜索流程,而pFind 3则是软件的一个版本。具体来说,Open-pFind这个开放式搜索流程,是pFind 3软件的主力流程。实际上,大家用pFind 3还是可以选择限定式模式的。
Open-pFind的研究大概是起于2013年8月,到2013年底,我完成了软件内核的研发,也就是现在算法的80%;后来若干年,则不断地进行结果验证、性能优化、开发配套软件等等,直到2016年底,开始写作第一版文章,2017年底投出(汗……写出来才发现这个时间段比我想象得要长), 2018年8月正式接收,大概有5年时间。而实际上,这项工作的起源更早一些,大概是从2011年初,我便开始了深度解析相关的工作,也开发了Open-pFind的前一代开放式搜索版本(pFind-Alioth)。所以,这篇文字我打算从 2011年开始写起,借机梳理一下我这八年来数据库搜索相关工作的脉络。
1. 深度解析起源与pFind-Alioth(2011)
2010年,我基本完成了我自以为可以够博士毕业的两项工作,一是pFind 2流程(2010年发表在RCM),二是从头测序算法pNovo(2010年发表于JPR),所以当时没有毕业压力(后来RCM文章因为是共一作,在计算所不作数,还需要再写一篇,这是后话),心态还是比较放松的。于是 2011年初,贺老师希望我做一下深度解析相关的课题,这是我们组的第二个“973”课题,而我们承担的任务,可以归结为一句话,就是“提升串联质谱图的解析率”。
要提升解析率,首先要分析未解析谱图到底是因为什么原因没有被解析。实际上,早在2006年前后,Nesvizhskii博士,以及UCSF的Chalkley博士等人,就发表了相关的文章,探索未解析谱图,但我们仍然希望拿到第一手的材料,有个直观感受。于是,我和我们组的硕士生陈海丰(也是pNovo+算法的作者),针对董老师实验室的8个标准蛋白数据进行标注。这批数据共包含 951个母离子,每个母离子分别打了两个HCD谱图、一个CID谱图和两个ETD谱图,我和海丰分头过每一张谱图,利用五张谱图的综合结果来认定母离子的身份。
当时的主要策略就是拼凑工具:pFind搜索得到基本结果àpNovo从头测序查验未解析谱图,或者使用pCluster、pMatch等工具进行修饰发现与鉴定à回贴至蛋白序列,并查看谱图匹配情况à看是否可解释为修饰、半酶切或突变à指定对应的精细参数,进一步搜索,得到更多结果。我记得这项工作大概耗时三个月左右,完成时是2011年的4月。最终结果是我们至少以当时的认识,把每一张谱图尽可能地给出一个身份。最后的分类如下图所示, 80%以上的谱图(指母离子)可以解析为肽段,而常规解析率只有50%左右。这项研究虽然非常繁琐耗时,但是最终给了我信心:谱图可利用的部分绝非当前这些,我们需要想办法做的,只是要把上述手工提升解析率的办法自动化。
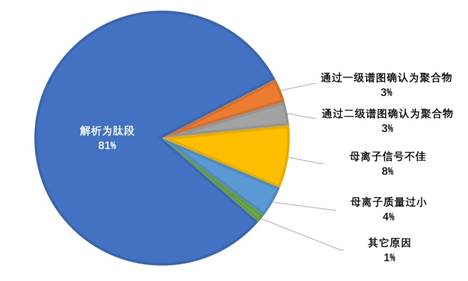
进一步,我又分析了腾冲嗜热菌的一组数据,含有6000多张谱图,也是采用同样的思路,解析率可以从40%提升到70%左右。重要的是,在这里,我发展了pFind-Alioth离子索引流程,自动化地解析含有未知修饰或者意外酶切的谱图。
我想还是有必要说一下pFind-Alioth相关的技术特点,因为它和后来的Open-pFind是有很多相似之处的,从纯流程设计角度讲,更是比后来Nesvizhskii团队发表在Nature Methods的MSFragger软件要更精巧一些,尤其是在处理非特异酶切肽段检索方面。
离子索引的大致思想是:对于每张谱图,其大概有100个信号谱峰,每个谱峰进行一次检索,记录一下匹配到了哪些肽段。全部检索完毕后,引擎会发现,对于肽段A,它匹配到了50个谱峰;对于肽段B,匹配到了20个……这就像是一个打分一样,一下子把匹配很好的(通常也是正确的)肽段凸显出来了。
这种方法之所以快,就是因为,它避免了“空匹配“:
传统引擎:我们可以想象,母离子质量容差由20 ppm扩增到500 Da,虽然候选肽多了很多,但正确的只有一个,剩下的都是陪太子读书,能有匹配上一两个离子就不错了。所以,传统方法进行开放式搜索,绝大多数候选肽与谱图打分都接近于0——然而,虽然结果是0,过程上可一点不省时间,因为也是完整的一次打分。
离子索引:每个离子去查询索引,只要检索到肽段,那就相当于在传统引擎中,实打实地贡献了打分。我们刚才说过,大部分肽段与谱图几乎没有什么匹配,所以,离子索引检索下来,能够有效匹配上的肽段(比如,匹配到的离子数目多于5个),是极其有限的。
这个方法,其实和谷歌检索的做法是一致的。我们可以想象,如果在谷歌输入一个关键词“蛋白质组学 CNCP”,它是不会对每个文档都检索一遍,然后看谁与这两个关键词最像,最后把所有网页排序输出的。相反,它会检索后台的索引,看看哪些匹配上了“蛋白质组学”,哪些匹配上了“CNCP”,哪些匹配上了这两个词,哪些匹配上了两个词且还不止一次。前面就是传统引擎的做法,后面就是离子索引的做法。
这项工作于2011年底投稿RECOMB CP会议,但是很遗憾被拒稿了。后来的2012年到2014年,我的工作重心先后放在pNovo+的研发、博士毕业与留所、 Open-pFind算法研究和pFind 3软件开发上,直到2015年初,我才把这项工作投出去,发表在Journal of Proteomics。晚是晚了点,但是我并不是太遗憾(当然如果它在我毕业前能够中稿,或许对我的毕业论文有所加成),因为我已经发现了更好的搜索策略,也就是后来的 Open-pFind。
2. Open-pFind的诞生(2013)
2013年6月,我历时七年拿到了博士学位。支持我毕业的两篇文章,其实都是和从头测序相关的;但是pFind-Alioth工作,却是我更看重的。当然,随着我在更多数据集上不断测试, pFind-Alioth,或者说,离子索引,存在两个比较大的问题,就是查询的特异性不是很高,以及占用内存空间很大。
关于查询特异性不高这个问题,简单说,就是给定一个待查询离子,比如,谱图中质量为300.4567 Da的一电荷谱峰,我们需要假设它为b离子或者y离子,然后,利用离子索引,查询数据库中存在哪些与其质量相匹配的理论碎片离子。这个数量是非常多的,比如,对于Yeast数据库,每个质量在300–1000 Da的谱峰可以查到上万个离子匹配。当然,对于一张谱图,我们会挑选多个谱峰进行查询,并查看哪些肽段结果对应的查询谱峰较多;但这无疑是增加了查询次数以及查询得到的理论离子数量,对检索效率有很大的负面影响。
关于占用内存的问题,我们可以估算一下。对于1M大小的蛋白质序列,碎片离子所含有的氨基酸片段假设不超过40,那么,考虑非特异性酶切下的y离子,共有40M个。每个离子需要存储其在蛋白质内的位置(4个字节),以及质量数(4个字节),至少也要8个字节。这样的话, 我们需要至少 320M空间存储这些理论离子。再加上一些附加信息,这个空间可能会大于500M。当时64位机器+64位操作系统并不像现在这样普及,所以,一个程序最大的可用内存,通常只有2G。而我们平日所用的数据库,基本都是在3–100 MB之间。所以,数据库建成的索引无法一次性导入内存,我们只有分批搜索。当然,如果只考虑特异性酶切,那么离子索引所占空间可下降一个量级;而且后来 64位机的出现,内存消耗从算法问题变成了经济问题。但在当时,我对离子索引的上述两大缺陷还是很不满意的,决心要修改一下。
那么,有无更好的查询方式呢?我把目光转向了序列标签(Sequence Tag)。的确,它可以很好地解决上述两方面问题。首先,它的特异性是极高的,尤其是在标签长度比较长的情况下。比如,还是针对Yeast数据库,绝大多数5-tag(就是含有5个氨基酸的序列标签)只匹配到不到10个蛋白质;换言之,只要你在谱图里提取到一个准确的 5-tag,那么你只需要对数据库里的10个“区域”进行候选肽段的生成及打分就可以了。另外,序列标签占用内存很少,同样对于1MB大小的数据库,理论上5-tag也只有1MB,大概用4MB内存就可以存储它们了(需要对标签进行编码压缩)。
所以,的确,序列标签索引看上去很美。当然,它也有它的问题,就是能快速、准确地提取到准确的标签,其实还是比较难的。好在我此前有从头测序的技术储备,所以做标签提取的工作还是相对顺利一些。另外一个难点,就是很多谱图其实本身就无法提取到长序列标签,这势必会造成某些中低质量(quality)谱图难以解析,所以解析率还是无法有效提升。针对这一点,我在开放式搜索之后,补充了一个限定式搜索的过程,利用限定式搜索来把开放式搜索中的漏网之鱼打捞起来,最终,形成了现在Open-pFind的两步搜索流程。
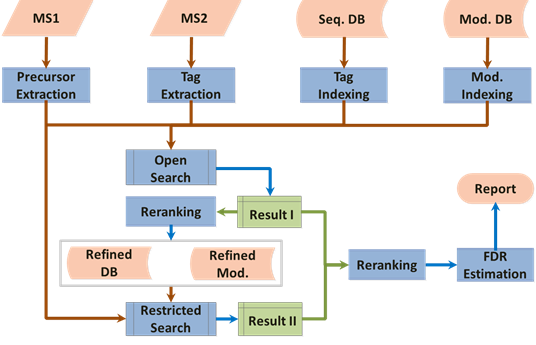
以这个流程为基础,Open-pFind相关工作为贺老师2013年的BCEIA会议报告添加了一些砖瓦。这也是令我印象很深刻的一次报告准备过程,因为在这个过程里,引擎的许多细节得以快速敲定,相当于是一次急行军。到2013年底,Open-pFind已经是一个比较完善的核心流程了。
3. Open-pFind的发展(2014 – 2016)
所以,Open-pFind其实在2013年底的时候,就已经开发得差不多了。那么,为什么到2016年我们才开始准备投稿呢?这中间,其实经历了相当长时间的优化改进和结果验证过程。一是要把引擎做得更快更准更全,即提升绝对性能;二是要充分说明我们的确做得更快更准更全,即构造评价体系。
我总结一下这几年的改进,如下:
Ø 基本评测,明确优势:要评测引擎,首先要选择数据集,以及搜索引擎,进行全面比较。在2014年底和2015年底,我在组内完成过两次技术报告,分别是与3 ~ 5种引擎,在4 ~ 6组数据集上,比较解析率、一致性和运行速度等指标。2016年底,开始在前面的基础上,确定了7种引擎(Open-pFind, PEAKS PTM, MODa, Comet, MS-GF+, Byonic和 pFind限定式搜索),在6组数据集上进行对比,开始进行文章写作。
Ø 发展软件,形成整车:2014年,我的主要工作是带领组内杨皓、瑞敏、刘超、邬龙等小伙伴,一起开发完整的pFind 3软件。整个集中开发历时半年多,后来则是一点一滴的版本改进。目前界面版本已经发展到3.1.4(2018年11月21日发布)。
Ø 他山之石,可以攻玉:15N标记数据一直是我们组内持续使用的结果验证手段之一。简单讲,就是给定多重标记样品(比如无标记,15N标记和13C标记),使用引擎进行无标记搜索;得到结果后,查看每个结果肽段在一级谱图中是否有对应的标记信号,作为鉴定结果是否可信的一个佐证。Open-pFind从2015年开始使用此方法进行系统评测,并在此过程中发现和修改了很多软件的 bug,优化了打分模型。刘超的pQuant,无疑是这一步的重要武器。杨皓在帮助我写完软件界面之后,也在此分析过程中出了不少力。
Ø 项目应用,以赛代练:2016年,复旦大学陆豪杰老师牵头,我们参与申请了国家重点研发计划重点专项。Open-pFind相关研究当时正在进行中,也成为第一课题的重点研究内容。在项目申请过程中,我们需要对人类蛋白质组草图工作进行系统评价,这也促使我使用当时的Open-pFind引擎,对整套数据集(包括2,212个raw文件,两千五百万张谱图)进行分析。在这个期间,我们也发展了一个临时的集群版,同时也对单机版的速度进行了最后一次比较大规模的优化。
时间到了2016年底,我开始了文章写作,目标是投稿Nature Biotechnology。坦白说,当时心里一点底都没有,因为作为相对不那么新的数据库搜索引擎技术来讲,MS-GF+于2014年发表于 NC,在我看来就已经是个突破了。后来Gygi的开放式搜索工作发表在NBT上,但纯技术层面来说没有太多新意,同时挤压了其它引擎的期刊发表空间。
不过不管如何,先试试看,即使被拒其实也就耽误一周。
4. 写作、投稿、修改和中稿(2017-2018)
写作过程没有此前想象得那么顺利。首先,对数据的重分析几乎耗费了两三个月,这期间我把Introduction和Results部分基本写好了。但是,2017年4月,MSFragger横空出世,我这里被迫对文章做比较大的调整,主要是需要跟它做全面的比较,从技术细节到结果分析和实验现象论证。MSFragger工作本身倒也没什么,我前面写过,其实我们 Open-pFind的工作相当于是pFind-Alioth离子索引工作的改进版,而MSFragger单说离子索引技术本身,还不如pFind-Alioth完整(当然,MSFragger的后续分析写得非常全面深入,值得学习)。但不管如何,相似工作发表给我这边带来的压力还是极大的。
直到6月份,我才把MSFragger相关的部分加入到文章中,随后写作Methods,调整附图附表等等,到10月份完成了初稿。后来两个月,贺老师、董老师、徐老师及各位作者帮忙做了很大的修改,另外我还请专业编辑润色了文字。 12月初,厦门质谱会议之前,成功投出。当然,仅有一点点不切实际的期待。
果然,在从厦门返程的高铁上,我收到了拒稿信。虽然没有期待,但难免有些失落。当然,化悲痛为力量,我还是跟大家打了一路扑克。回来之后,我跟贺老师商量,打算再尝试Nature Methods。不过贺老师说,还是可以尝试写一点rebuttal。我一开始内心其实是拒绝的,因为总感觉作用不大,但是还是去尝试了。可以申诉的点只有一句,就是编辑认为我们的工作“too incremental”。但是在写作邮件的过程中,我突然觉得,我们其实是有充足的理由可以申诉的,因为作为开放式引擎,我们的测试结果表明,Open-pFind不是简简单单的性能提升,这只是表面现象,只是最终的一个效果展示而已;深层次的话,从多个技术环节我们是有明确创新的,这个创新点需要我们明确提出来。于是,我真的是越写越有信心,写完之后,我自己觉得我作为编辑都肯定都要被感动了 :-)。
邮件发出后,当然是例行地作为低优先级稿件被编辑处理。而且,当时正赶上圣诞节和新年,编辑问我是否要等。我想,反正我也不靠这个毕业了,等!这一下就从12月中旬等到了2018年1月下旬。1月26日,收到了编辑的回复:“I'm very pleased to inform you that we have decided to overturn our decision and the manuscript has been selected for peer review.”。我觉得,可能是他们觉得让我等了一个多月,有点不好意思?当然,不管如何,还是送审了,对我而言,是历史性突破。
3月16日,收到第一轮审稿结果。三位评审人,第一位给了最多的意见,但对文章仍然是个偏积极的态度;第二位评审人持负面意见,认为创新性不足,建议投稿Nature Methods;第三位评审人的意见最为正面,提出的意见也是好奇多于质疑。具体大家可以查看本文附件给出的文章评审意见。另外,编辑根据第一个评审人的意见,建议我们把文章类型从 Article修改为Brief Communications。
4月25日,我们返回了第一版修改稿。最大的补充实验,是额外使用陷阱库策略,对软件进行了独立于目标-诱饵库、代谢标记这两种方法的第三种性能评测。虽然所有问题都已经回答,且针对部分问题做了超量的工作,连问题带答案大概有 38页,但这时候心里仍然没有底。
5月18日,收到第二轮审稿结果。编辑只给了第一位和第三位评审人。第三位评审人基本满意了,而且他有些为我们争取发表一篇Article的意思;但是编辑依然是维持此前的决定。第一位评审人对修改比较满意,但还是提出了不少细节问题。最大的一个问题是, pFind搜索标记数据过程中,会不会把某些重标母离子当成轻标进行搜索,还得到了(错误的)肽段结果并报告出来呢?这个问题的确问得有道理,我进一步补充实验,最终证明此前搜到的结果里面,最终仅有不到1%的结果,可以被pFind搜索到一个更好的重标肽段;而且,这里面还有相当多的谱图属于混合谱,意味着轻重标结果可能都是正确的。
5月29日,提交第二版修改稿,文字量大概是第一版的三分之一。
6月26日,Accepted in principle,这一天也是我博士毕业五周年。
7月8日,根据编辑意见返回修改稿;7月16日和7月 18日分别又做了一些小改动。
8月3日,正式接收。10月8日在线发表。
5. 小结与感想
感谢:真的非常感谢董梦秋老师、徐平老师和张佩珩老师团队。这篇文章中的E. coli轻重标1:1:1数据集是董老师实验室制备的,是阐述Open-pFind准确度的最重要一环。徐老师的酵母轻重标1:1数据集同样也用来说明准确度,且在阐述速度优势时有独特作用。人类蛋白质组大规模质谱数据Kim Data是在张老师的生物信息数据分析存储一体机上跑的,仅用时 5个小时就将两千五百万张谱图搜索完毕。在速度优化过程中也得到了谭光明老师和刘涛老师的诸多帮助。此外,文章发表前后,得到了很多祝贺;我个人状态反复的时候,也得到了很多人的关心;更重要的,大家在使用软件过程中,给了很多反馈,帮助我们不断前进。无法一一感谢,权且在这里统一表达我的感激之情。当然,还有贺老师、孙老师、刘超、杨皓等我们组的众多pFinder,他们陪我度过了太多难忘的美好时光。
未来:Open-pFind的发表,只是算法研究方面告一段落(实际上,仍然在持续优化当中)。在这之上,还有软件开放后的一系列更艰难,也更有用的的事情。这可能是我未来需要着重投入的一点。当然,由于身在科研岗位,还有很多其他事情需要处理,一些软件需求不一定能在第一时间响应,所以也请大家多担待,我们会努力做下去,然后,总会保持进步。
花絮:我回想了一下这项工作历程中的关键时刻,想到很多场景,但在这里只分享一个我觉得蛮值得纪念的技术突破时刻: 2011年8月31日下午,改进了pFind-Alioth某个重打分算法,然后看着每张谱图输出的结果里面,我期望的高丰度修饰结果都乖乖地跑到前面去了,看到谱图解析率突然猛增了10个百分点。
查了查,似乎当时还顺手发了一条微博。现在看到后,除了感慨往昔峥嵘岁月,第一反应是我居然工作时间发微博……果然不同时间段心态也不太一样啊。
最后拿贺老师常说的话来收个尾吧:念念不忘,必有回响。祝福CNCPer,祝福中国的蛋白质组学团队,未来必定会收获满满!
2018年12月3日

我的第一篇科研论文是怎样诞生的——pLink 2研究历程回顾
陈镇霖
2012年7月,正在武汉大学读书的我,并不知道交联质谱领域正有一个重要事件发生——北京生命科学研究所的董梦秋老师团队和中科院计算所贺思敏老师的pFind团队合作研究的交联质谱鉴定软件 pLink发表在Nature Methods。为什么说“重要”呢?第一,这是大陆蛋白质组学研究团队首次突破Nature子刊,而且后续研发的二硫键鉴定软件pLink-SS于2015年再次发表在Nature Methods(两个软件合称pLink 1)。第二,交联质谱技术在蛋白质结构建模和相互作用研究中相比传统技术具有独特优势,所以后来pLink 1软件被近千位国内外用户下载,助力上百篇科研论文发表,其中有 40%的文章发表在CNS (Cell、Nature、Science)正刊或子刊,包括大神施一公和颜宁的几篇大作。第三,当我从武汉大学毕业、投到贺老师门下、加入pFind团队之后,居然成为了pLink的接班人!
科研无止境。pLink开局不错,但有很大的提升空间,比如搜索速度相对较慢、精度评价指标相对单一、软件易用性不足甚至很差。为此,2012年pLink文章发表之后,孟佳明师兄开启了pLink 2的研究工作。佳明师兄借鉴迟浩师兄 pFind-Alioth的思路,将碎片离子索引技术迁移到交联肽段鉴定中来,使得pLink 2的搜索速度相比于pLink 1有了很大的提升。此外,佳明师兄重新编写了pLink 2代码,高效的新版代码为后续的研究与开发奠定了坚实的基础。2014年,佳明师兄毕业,尹吉澧师姐接力继续改进 pLink 2,重点优化了pLink 2的半监督机器学习重打分算法,稳定提升了pLink 2的灵敏度。2016年,吉澧师姐毕业,我进入实验室,继续改进pLink 2。所以,这篇文字从2016年开始写起,主要介绍我接手 pLink 2三年半来的“苦难”历程。
1. 传承接力,评测先行(2016)
2016年上半年,吉澧师姐即将毕业,组里安排我接手pLink 2。那时候我正在雁栖湖校区学习(京区中科院研究生研一都要在雁栖湖校区集中上课)。为了更好地接手和学习pLink 2,我调整了课程表,每周空出一天从雁栖湖回到计算所,向吉澧师姐学习pLink 2。半年的时间,前后共交接了13次,回想起来,每次早晨6点迎着朝阳出发,下午5点伴着夕阳返回,别有一种“日出而作,日落而归”的感觉。那时的我,不太了解pLink的辉煌历史,也不会想到 pLink 2研究工作的复杂性,只是按部就班地完成组里交给我的任务。
吉澧师姐给我安排的交接任务循序渐进,首先阅读师兄师姐们的毕业论文,然后学习如何使用pLink软件,最后才深入到代码细节。交接之余,师姐还会给我介绍pLink的历史以及pLink未来可改进的方向。师姐耐心的传授将我带入了交联质谱鉴定这一领域,以后每年我给刚入组的新生培训pLink内容时,都会想起当初吉澧师姐与我交接的情景。交接pLink代码期间,也是我第一次接触一个庞大的 C++工程项目。pLink 2内核共有100多个源文件,总计3万多行C++代码,一次完整编译需要将近5分钟,这是我在本科期间从来没有接触过的大型项目。虽然代码量很大,但理解起来并不费劲,这一方面得益于吉澧师姐的讲解,另一方面得益于佳明师兄最初良好的编程风格。佳明师兄高质量的代码实现为后续的算法更新和功能拓展提供了很大的便利。经过半年时间的交接学习,我对 pLink 2的算法实现和软件使用有了宏观上的掌握,并且能辅助师姐修改一些小bug(软件错误)。
2016年下半年,吉澧师姐毕业,我回到计算所开始了正式的科研之路。由于pLink 2历经两届学生依然未了,且遭到了当时发表的另一交联肽段鉴定引擎Kojak的挑战,贺老师决定先对pLink 2和Kojak进行全面系统的评测,摸清pLink 2的优势与不足,以便指导下一步工作。我参考师兄师姐们的毕业论文,开始对两个软件进行密集的评测。从 9月入学到10月,前后一个多月的时间,使用了四种不同的评测方法,写了四份不同的评测报告,平均下来每周要完成一种评测、写作一份报告并修改上周的报告。那段时间是我研究生阶段第一次感受到了巨大的科研压力。通常低年级学生进入实验室后都由高年级师兄师姐指导,但pLink方向的师兄师姐恰好都毕业了,我需要直接向贺老师汇报,这又无形中增大了压力。我至今仍然清楚地记得,贺老师对我的合成肽段数据集评测报告进行了严厉的批评,认为我低估了工作的复杂性,作为刚入组的萌新,我真的吓坏了。刚进入实验室的那半年,我一共写了 8份技术报告、共计19个版本,贺老师修改了8份报告、12个版本。虽然那半年做实验、写报告、改报告的过程很痛苦,不过也正是通过这一系列实验与报告,我基本掌握了后来pLink 2文章中的四种评测方法,也摸清了pLink 2的精度优势和速度不足。
经过评测,我发现pLink 2的速度虽然相比于pLink 1快了数十倍,但相比于Kojak优势不足,甚至在有些数据集上比Kojak慢,因此下一步的目标就是对pLink 2进行加速。速度优化是一个定义明确的问题,我借助热点分析工具将性能瓶颈确定在粗打分环节,通过减少谱峰的冗余排序、优化粗打分代码实现和引入位向量技术,我将 pLink 2的速度由原来比Kojak慢2倍提升到比Kojak快3倍的水平。以此内容为基础,我完成了2016年的年终技术报告——这是我2016年的第 8份技术报告。
2. 算法软件,厅堂厨房(2017~2018)
优化算法
2017年,我进一步扩大了评测范围,增加了评测数据集的多样性,并在董梦秋老师实验室进行了用户场景下的测试。大多数情况下,pLink 2的精度优势依然保持得很好,但在两种情况下pLink 2的灵敏度异常低。
第一种情况是,pLink 2在某些数据集上过滤出来的交联谱图数目只有个位数甚至为零,但pLink 1能鉴定到几百张交联谱图。凭直觉我以为这是pLink 2的小bug,但经过仔细排查,发现是pLink 2的半监督机器学习重打分算法由于正样本不足而无法在线训练,而正样本不足的原因又是因为重打分算法冷启动时使用的细打分算法其谱间归一化效果较差。明确问题之后,我提出了“离线训练 +在线训练”的重打分模式,即首先离线训练一个机器学习重打分模型,然后在线训练一个机器学习重打分模型;当在线训练由于正样本不足而无法进行时,调用离线模型进行第一轮打分;如果第一轮打分后能获得足够数量的正样本,则进入在线训练,否则直接使用离线模型的结果。“离线训练+在线训练”的重打分模式一方面为在线训练提供了较好的初始正负样本,有助于训练更快收敛,另一方面即使依然无法进行在线训练,离线训练所得模型的打分也比使用细打分的谱间归一化效果好很多。因此,“离线训练 +在线训练”的重打分模式极大地提升了pLink 2软件的鲁棒性,消除了灵敏度异常的现象。
第二种情况是,pLink 2在面对较多可变修饰时,灵敏度显著低于平均水平。这个现象是我在分析Kojak论文数据集时发现的,该数据集要求添加五个可变修饰,而常规交联搜索流程一般只设置一个氧化作为可变修饰。经过分析,我发现离子索引设计之初对可变修饰的考虑不够充分,导致一条肽段的多种修饰形式的匹配峰计数无法区分,进而导致修饰肽段的匹配峰计数偏高。而随机匹配的肽段往往带有较多的可变修饰,这就使得随机匹配肽段打分偏高,导致算法整体灵敏度偏低。为了解决这个问题,我对离子索引的结构进行了修正,新增了 10比特用于区分同一肽段的不同修饰形式(最多支持210=1024种修饰形式),这就是最终pLink 2文章中的离子索引结构。
除了上述两个带有研究性质的问题之外,大规模的评测也发现了一些小bug,比如I/O耗时较长、谱图预处理时电荷判断不准等,也都采取了相应措施予以修复。经过一年多的评测、优化、再评测、再优化,pLink 2的内部算法趋于稳定。
发布软件
2017年下半年,贺老师决定于2018年元旦面向全球用户发布pLink 2软件。做出这个决定其实很难,贺老师和我都有过纠结,因为软件发布前后涉及到大量的软件测试、bug修复和用户支持等工作,势必影响文章写作和投稿的进度。虽然如此,贺老师觉得软件相比于文章更加重要,而且从长远来看,软件的先行发布对后续的文章写作和投稿也有帮助。贺老师一直强调生物信息学工具不但要做到内部算法漂亮、彰显专业智慧,还要做到软件能用、好用,即所谓“上得厅堂、下得厨房”。为了此次软件发布,我在软件易用性方面又下了不少功夫,如新增了 E-value计算功能,整合了pQuant定量模块,重新编写了界面代码,制作了用户手册等。特别地,我还精心为用户准备了新年问候信,并请董老师帮忙润色,那也是我第一次感受到董老师如丝般顺滑的英语,写得又漂亮又暖心。在12月临近发布时,我动员了全组同学帮忙测试新版pLink 2软件,曾文锋师兄是当时软件发布的总指挥,我至今还记得在 2018年元旦前夕,文锋师兄和我一起熬夜准备软件发布前的一切事宜。那个时候真真切切地感受到了互联网公司在新版软件上线前的紧张和忙碌。
虽然软件发布前我们进行了大量的测试,但当软件对外发布后,小问题依然不断。特别是pLink的上千个用户遍布世界各地,每个用户使用的操作系统和软件环境千差万别,明明在实验室测试一切正常的功能,在用户环境下就是崩溃。令我印象深刻的一个问题是,英国之外欧洲国家的小数点不是点号“.”而是逗号“,”,导致pLink 2读取用户设置的错误率FDR阈值有误(如将 0,05解析成了0)。由于我并不知道这一“常识”,在软件编写时自然就无从考虑。此后,为了保证软件发布前的测试足够充分,我在虚拟机中安装了3种不同语言(中文、英文、法文)的3个不同的Windows版本(Win7/8/10),每次发布前,都要逐个测试,确保都没问题之后才上线。
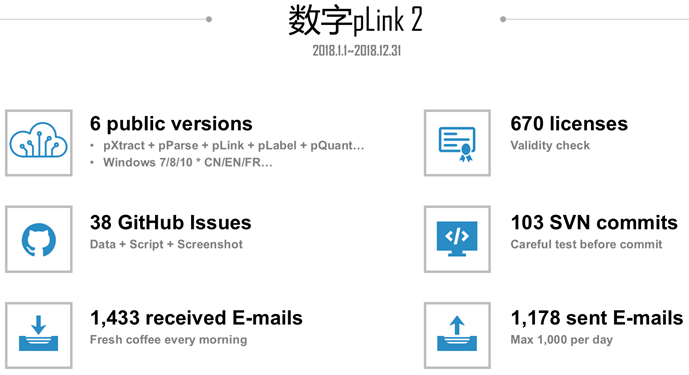
2017年和2018年在软件维护和推广上花了不少时间,下图是2018年1月至12月的工作量统计数据。仅从 pLink@ict.ac.cn发送的邮件数量来看,如果认为发送pLink许可证(License)是一件不需要消耗任何时间和精力的事情,则我发送了1178 - 670 = 508 封需要消耗时间和精力的邮件。一年去掉寒暑假和周末的时间,工作日大约有220天,则平均每天需要给用户发送2.3封邮件。这2.3封邮件中,有些可能几句话就能说清楚,也就是十分钟的事情,但是有些问题需要帮用户跑软件、分析数据,需要写详细的过程,少则一个小时,多则半天一天。再加上GitHub上的 38个提问(Issue),大多数提问的回复都很详细,而且为了让用户便于理解,有些还需要图文并茂。平时还有一些用户会通过QQ、微信等途径进行咨询。遇到年中、年末发布新版本时,各个模块的更新、测试更是一个浩大的工程,那段时间往往连续一两周都在忙软件发布的事情。所以保守估计,平均每天用于维护软件、客户答疑的时间需要2个小时。处理 pLink邮件成了我每天早上的提神“咖啡”(Fresh coffee every morning),我常常早上来到实验室之后,优先处理pLink邮件,不知不觉一上午就结束了。
功夫不负有心人,pLink 2仅仅发布一年半的时间,其注册下载量就超过了pLink 1七年注册下载量的总和。更为重要的是,在迄今为止的一年半当中,已经有8篇高影响因子文章明确使用而不是简单引用了pLink 2软件,其中包括 3篇CNS子刊和1篇PNAS文章。pLink 2软件的先行发布,也无形中为后续pLink 2文章的审稿增添了积极因素。我们在每次提交文章初稿/修改稿时,都会在投稿信即 Cover letter中与编辑分享pLink 2软件的最新应用成果,我相信编辑看到这些成果时,会更加积极地考虑我们的文章。
2017年还发生了一件大事。那年秋天,我升入硕士三年级,同时在校招季拿到了不少互联网公司的录用通知书。我和大多数硕士学生一样,等着第二年毕业进入公司上班。也许是冥冥之中自有定数,某一天的午后,我偶然看到了卡内基梅隆大学李沐博士的一篇博客《博士这五年》,这篇博客回顾了李沐读博的心路历程,其中有句话深深地触动了我:“更重要的是理想和情怀。人一生要工作五十年,为什么不花五年来追求下理想和情怀呢?”想到已经是硕士三年级的我,马上要毕业了但依然没有拿得出手的科研成果;想到工作什么时候都可以找,但如果就此毕业,以后可能就很难有继续读博与科研的机会了;想到pLink 2文章依然悬而未决,我毕业之后文章何时才能发表……那个下午,我想了很多很多,一个从来没有过的想法逐渐在脑海中清晰起来——我要读博,为了理想和情怀。我与贺老师沟通了这个想法,贺老师很支持我读博,也尊重我的选择。那年国庆,我给自己放了假,出去玩了一整个假期,回来之后,我正式决定转博,继续推进pLink 2的研究工作。
3. 系统评测,不断深入(2018)
转博之后,我一边准备pLink 2软件发布,一边着手pLink 2论文实验。我将进入课题组以来做过的所有评测实验进行总结,归纳出四种评测方法,从易到难分别是模拟数据集、合成肽段数据集、15N标记数据集和陷阱库方法。使用这四种方法对Kojak、pLink 1和pLink 2三个引擎在化学交联数据集上进行了系统的性能评测。值得一提的是,尽管大多数交联鉴定引擎会使用蛋白质晶体结构信息来检验交联鉴定结果的可靠性,但我们从一开始就没打算使用这一方法。因为晶体结构记录的是蛋白质的凝聚状态,无法客观反映蛋白质在溶液中的动态构象,董老师此前的研究证实了这一点。使用晶体结构来检验质谱鉴定结果,相当于用冰的沉稳预测水的灵动,是相对间接和粗略的证据。而pLink 2文中的四种评测方法都没有脱离质谱鉴定,比晶体结构检验更加直接和精细。
2018年1月份,经过一个月左右的集中写作,我完成了pLink 2论文的第一份英文初稿。现在看来,虽然那个版本中正文的几张图已经和最终发表的文章中的图很接近了,但文章内容非常单薄,不但评测的广度不足,评测的深度也不够。不过当时我并不这样看。
2018年上半年,贺老师对我的第一份英文初稿批注了大量意见,要求对文章内容进行大幅扩充,包括评测更多的交联引擎、每种评测方法同步增加二硫键数据集、充分论证离子索引加速开放式搜索的过程、分析15N标记检验的错报率和漏报率等,既涉及评测广度,也涉及评测深度。看到这么多批注意见时,我的内心是崩溃的,我当时想为什么不能只用现有的素材投个J Proteome Research呢。抱怨归抱怨,我还是硬着头皮去做了,由此开始了我“苦难”的2018年。
增加评测广度
在评测广度方面,我不但增加了参与评测的引擎数量,还增加了参与评测的数据集数量。我阅读了大量的文献,选择了当时主流的9个交联鉴定引擎,逐一下载试用。这个过程是漫长而乏味的,每个引擎的使用方法千差万别,各种特有的参数五花八门。通常,运行一个软件是很难一次性成功的,遇到崩溃的情况还需要查软件的使用手册,甚至发邮件给软件作者确认使用方法是否正确。经过一个多月的时间,我基本摸清了每个软件的使用方法,并获得了它们在化学交联数据集上的评测结果。
由于pLink 1软件还集成了二硫键鉴定软件pLink-SS,pLink 2如果想全面替代pLink 1,还需要补充二硫键数据集的相关评测。当时实验室的方润乾师弟的课题和二硫键鉴定有关,在这个紧要关头,润乾也帮我分担了很大一部分的二硫键数据集的评测实验。在二硫键数据集的评测当中,很值得一提的是15N标记的E. coli样品。一开始,我们发现几个交联引擎的鉴定结果的定量比值异常比例都很高(高达30%),远远高于之前做过的标记样品的定量比值异常比例(通常<5%)。更为奇怪的是,即使相对可信的多引擎鉴定交集结果,其定量比值异常比例也依然很高。后来,董老师的博士生曹勇师兄分析发现,这些定量比值异常谱图主要来自两条和金属锌Zn相关的蛋白,于是怀疑样品在标记时受到了重金属污染。再后来,我们通过测定样品中的微量元素含量确认了此事,并通过更换试剂公司成功解决了这个问题。虽然这个数据集的评测结果只放在了附录当中,但其背后的故事是曲折的。
增加评测深度
在评测深度方面,我对2016年刚入组时做的每种评测实验都进行了更深入的分析,下面重点介绍在模拟数据集和15N标记数据集上的深度评测。
模拟数据集作为一种无需湿实验即可生成的数据集,几乎可以无成本地模拟不同数据规模和数据质量,是贺老师在组内倡导的一项通用技术。我本以为这只是一项例行公事,所有引擎在高质量的模拟数据集上性能都会很好,没想到结果大大出乎我的意料,不同引擎的性能差别很大。比如搜索速度方面,在第一版手稿中,我使用E. coli数据库中的500个蛋白随机生成一万张模拟谱图的数据集,pLink 2只用1分钟即完成搜索,即使是pLink 1耗时也不到半个小时,但Xilmass和StavroX运行了一周还没有完成搜索;后来我不得不把数据库的规模从 500个蛋白缩小到100,这两个引擎才能在可忍受的时间内搜索完毕。多引擎的灵敏度差别也很大,9个引擎只有了pLink 2、pLink 1和Kojak的灵敏度超过85%,其他引擎的灵敏度都不足 80%,有的甚至不足50%。这一度让我怀疑生成的模拟谱图质量太差了,导致其他引擎难以鉴定。
我后来专门分析过StavroX和MetaMorpheusXL的灵敏度低的原因,发现前者需要用同位素峰簇来判断谱峰价态,如果只模拟生成碎片离子的单同位素峰而不生成其同位素峰簇,则StavroX的灵敏度会很低;后者对谱峰缺失很敏感,缺失的谱峰稍微多一点,则灵敏度下降比较明显,猜测可能和 MetaMorpheusXL使用离子索引但打分算法不够鲁棒有关。为了降低模拟数据集评测的难度,我参考合成肽段谱图的缺峰概率,模拟了一个很低的缺峰概率,并且生成了每根单同位素峰相关的同位素峰簇。在这种比较完美的模拟条件下,大多数引擎的灵敏度依然比较低。考虑到后续评测使用的是真实谱图集,且数据库规模大很多,很多引擎的表现可能不会太好,于是决定用模拟数据集的评测作为一个初筛,只选择性能较好的pLink 2、 pLink 1和Kojak进入后续的评测。
利用15N标记数据集来检验鉴定结果的正确性,也是贺老师在组内大力倡导的一项通用技术,此前在完整糖肽鉴定软件pGlyco 2的Nature Communications文章、常规肽段开放式鉴定软件 pFind 3的Nature Biotechnology文章中显过身手。其原理并不复杂,就是对引擎鉴定到的轻标结果进行定量,如果轻标信号在一级质谱图上能找到对应的重标信号,则可计算出定量比值,认为检验通过、鉴定不可疑,否则定量比值计算异常,认为检验未通过、鉴定很可疑(极大可能是错误的)。在一个15N标记化学交联数据集上,pLink 2鉴定谱图5196(其中定量比值计算异常比例0.5%),pLink 1为4774(3.8%),Kojak为2672(6.4%),表明 pLink 2具有显著的灵敏度和准确度优势。
但是整个标记检验涉及到多个环节,如前期的样品制备、中间的交联引擎鉴定和后期的pQuant软件定量,每个环节都可能引入偏差而导致定量比值异常。为了排除样品制备和软件定量的偏差,我先使用已发表的Open-pFind软件鉴定上述交联样品中的单肽,然后使用pQuant进行定量。结果表明,单肽鉴定结果的定量比值异常比例非常低(0.3%),说明样品制备和pQuant定量基本没问题,即使有问题,最多引入 0.3%的定量比值异常比例。如果后续换用交联引擎之后,定量比值异常比例远大于0.3%,则说明这是由交联引擎的错误鉴定导致的。
当然,15N标记检验技术本身并非完美无缺,通过检验不代表结果一定正确,有可能错误肽段和正确肽段的N元素数目刚好相同,导致15N检验出现漏报。类似地,没有通过检验也不代表结果一定错误,的确发现有可信的轻标鉴定结果没有检测到重标信号,导致15N检验出现错报。只有同时考虑漏报率和错报率,才能把定量比值异常比例换算为真实错误率估计。此前pGlyco 2一文假设错报率为0,pFind 3一文对漏报率估计偏乐观,之后周文婧师姐的pValid文章对相关理论和操作做了更一般的研究,我把文婧师姐相关工作迁移到pLink 2文中,对于15N标记检验的错报率、漏报率估计更加全面、更加谨慎。理论推导和实际计算表明,错误率和定量比值异常比例正相关,定量比值异常比例高,则所估计的错误率也高,因此引擎相对优劣的结论不变,但是整个论证更加严谨。不过,为了提高论文的易读性,我把更容易理解的定量比值异常比例放在了正文,而把换算后的错误率估计放在了附录。
半路杀出程咬金
当各种评测实验补充得差不多之后,我再一次燃起了投稿的信心。就在此时,我得到了两个令我不安的消息:MetaMorpheusXL和Xolik正式发表了(8月)。前者使用了离子索引,部分地削弱了我们文章的创新性;后者虽然没有使用离子索引,但使用了另外的打分模式和数据结构,使得速度比pLink 2还稍快一点,而且还号称灵敏度更高。
为此,我被迫对文章进行调整,新增与这两个引擎的比较,全文的评测引擎新增到了10个。更进一步,为了回应各个引擎对pLink提出的挑战,贺老师提出不但要用自己的数据集进行评测,还要重新分析和pLink对比过的文章的数据集,比如Kojak、Protein Prospector和Xolik。这样,我又花了一个多月的时间补充评测,结果表明: Kojak的挑战,属于对方参数设置不当,pLink依然具有全部优势;Xolik的挑战,属于pLink代码bug,改正后pLink精度依然有很大优势;Protein Prospector的挑战,虽有道理,却也不改变相对优劣。
特别地,在与Protein Prospector的对比评测中,针对其文章发现的对蛋白间和蛋白内交联结果分开过滤有助于提升蛋白间交联结果的可靠性这一现象,我不但从理论上进行了严格的推导,而且补充实验验证了我的结论。此外,我的理论推导还发现分开过滤不但有助于提升蛋白间交联结果的可靠性,而且有助于提升蛋白内交联结果的灵敏度。有趣的是,虽然分开过滤很有道理,但是用户反映, pLink合并过滤的很多蛋白间交联结果尽管不在分开过滤结果内,却依然很可信,所以要求支持合并过滤。于是我既增加了分开过滤的选项,又保留了合并过滤的选项。
总之,pLink 2通过了所有的考验,新增的评测也进一步充实了论文的工作量。
回顾整个2018年所做的评测工作,其实增加评测广度和增加评测深度是相辅相成、交替进行的;当评测的广度越大,发现的问题就越多,这驱使我分析其中的原因,增加评测的深度,进而把个别问题下的深度评测推广到更大范围。通过这一系列评测,不但能发现各个软件不同的异常现象,指导软件优化,而且能发现交联质谱鉴定的一般规律,加深对相关问题的认识,我想这大概就是系统性评测的意义。
4. 写作投稿,终成正果(2018~2019)
事实上,在2018年,软件评测和论文写作是交替进行且不断深入的,往往是补充完实验后修改论文,然后请贺老师批注,收到批注意见后又补充新的实验并修改论文,如此循环往复。在论文写作方面,由于缺乏英文学术论文写作经验,我参加了所里的学术论文写作研讨班,并重点学习了Nature子刊上发表的几篇交联引擎文章(比如刘凡博士的XlinkX文章)的写作方法。同时,2018年迟浩师兄的Open-pFind文章正处于审稿期间,Open-pFind手稿成了我非常重要的参考范本,比如pLink 2流程图的作图风格就和 Open-pFind很相似。
论文修改与润色
回顾pLink 2论文的写作过程,我个人觉得Methods和Results相对好写,因为实验方法和结果都由自己亲手完成,非常熟悉,只需将相关内容整理成英文即可。而Introduction和Discussion的写作相对较难: Introduction要求作者对领域有高屋建瓴的认识,不但能综述领域中有代表性的工作,还能一针见血地指出现存的问题,并自然而然地引出本文的创新点和主要成果;Discussion则要求作者客观公正地讨论本文的贡献和局限性,并前瞻性地指出领域下一步的发展方向。
作为科研小白,我毫无意外地在Introduction和Discussion的写作中栽了跟头。2018年年中,经过半年的补充实验,论文的Methods和Results逐渐丰满成型,但当我把修改好的论文手稿发给贺老师后,收到的批注中没有丝毫的表扬,相反,贺老师对我的 Introduction和Discussion非常不满。我至今还记得,在一个炎热的午后,贺老师把我叫到办公室,严厉地批评我Introduction中不先讲交联质谱技术对蛋白质结构和相互作用研究的突出贡献,而只讲其当前不足,很不明智;并语重心长地教导我Discussion不能草草了事,要有勇气指出当前领域存在的问题以及可能的发展方向,不要让读者以为 pLink 2为交联质谱鉴定画上句号。那个时候,我的心情低到了谷底,觉得自己好难,为什么连Introduction和Discussion都不放过。带着贺老师的批评以及沮丧的心情,我回家了,因为所里开始放高温假了(7月)。
在家的两周时间,我再次发扬了打不死的小强精神,把100多篇pLink相关的应用文章全部看了一遍,并根据不同的应用场景分门别类,然后总结成一小段突出交联质谱技术对蛋白质结构和相互作用研究的贡献的文字,贺老师看后大为欣赏。虽然后来经过多次压缩之后,这段文字变成了Introduction中的一小句话,但我觉得那两周高温假的努力没有白费。此外,我再次梳理了全文的创新点(离子索引加速和系统性评测的重要性),反复思考如何根据当前领域的问题一步步引出本文的创新点。我不断地理清思路、打磨文字,再通过贺老师的修改,几个来回之后, Introduction也变得有模有样了。类似地,Discussion也经过了多个来回的修改,不仅补充实验说明了序列标签即Tag索引对进一步加速交联肽段鉴定的潜能,而且还以谱图解析率和鉴定错误率为证据指出交联质谱鉴定依然面临技术挑战,并非pLink 2发表之后就万事无忧。
经过不断的补充实验,论文的工作量基本够了,但随之而来的是正文变得臃肿。于是,从12月开始,我们集中对正文进行了压缩和润色。我们请北京生命科学研究所的外教John Hugh Snyder帮忙对文章进行修改,John对文章进行了逐字逐句的修改,内容涉及常规英文写作中需要注意的语法问题,以及英语科技论文写作特有的连贯性、一致性等问题。由于文章和多个引擎进行了对比,如何客观评价其他引擎的性能又不至于伤害同行感情,我一直拿捏不准,董老师帮忙修改了部分语句,使得语气低调柔和了一些。其他作者也都对论文修改提出了宝贵的建议,无法一一详述。
在投稿期刊选择上,我们内部也有过不同的意见。开始时考虑Nature Methods,毕竟pLink和pLink-SS都曾发表在该期刊,但也考虑Nature Methods即使送审,很可能要压缩为短文(Brief Communications),不仅需要在文字写作上耗费大量精力,而且无法在正文中充分展开论述。我希望论文能尽快投稿、送审、发表,于是建议投稿Nature Communications,经过讨论,大家同意了我的建议。最终,历经一年多的写作和修改,我们在12月23日将论文投到了Nature Communications。作为一个科研萌新,怀着对Nature子刊的敬仰,我将当时的投稿过程录屏了下来:-)。
顺利的审稿过程
由于论文投稿期间正值国外圣诞和元旦假期,一直等到2019年1月15日才发现状态更新为“Manuscript under consideration”,我在心里给自己打气:“嗯,编辑送审了,加油!”
2月19日,收到第一轮审稿结果。两位评审人,第一位态度积极,肯定了系统性评测的重要性,但认为需要补充更多引擎在合成肽段数据集上的评测,增加引擎XlinkX的评测;第二位评审人持负面意见,认为文章过于计算,建议转投Bioinformatics,另外建议增加引擎 MassSpecStudio的评测。具体内容大家可以查看在线发表的全部评审意见及其回复。幸运的是,编辑的态度比较积极,给了我们修改的机会。
4月27日,我们返回了第一次修改稿。修改稿补充了大量的评测实验。首先是完善了模拟数据集的评测。为了说明模拟数据集的谱图质量很好,足以作为其他评测的基准测试,我参考合成肽段的谱图质量,使生成的模拟谱图比合成肽段的谱图质量还要好;另外,为了表明模拟数据集的公平公正,我在附录中详细描述了模拟数据集的生成方法,并且公开了相关代码。然后是补齐了所有 10个引擎在合成肽段数据集上的评测,评测结果进一步印证了模拟数据集上的结论,即性能排名前三的引擎依次是pLink 2、pLink 1和Kojak。最后是增加了pLink 2和其他两个引擎XlinkX和 MassSpecStudio的性能对比评测,结论再次表明 pLink 2具有很大的性能优势,这样评测的引擎总数增加到12个。
5月31日,收到第二轮审稿结果。那天早晨,就在我去领签证准备第二天出国参加美国质谱大会ASMS的路上,收到了Final revisions的邮件。作为没有投过文章的科研萌新,我当时并不知道 Final revisions意味着什么。当得知这是文章即将被录用的消息时,我甚至都不太敢相信。这一轮审稿结果没有特别大的修改意见,但建议我们谨慎处理对同行工作的评价,修改相关的文字。
6月15日,按照编辑意见返回修改稿。Final revisions给了两周的修改时间,正常情况下是够的,但6月第一周我正好在参加 ASMS,对于改文章的事情实在是有心无力,于是申请延期一周。幸好第二周回国之后快马加鞭按期修改完毕。
6月21日,正式接收。7月30日,文章上线。从2018年初写作第一个版本,到论文正式发表,前后共修改了 50个版本。
5. 感恩过去,砥砺前行
pLink 2的问世离不开pFind实验室师生的长期积累。pLink 2的离子索引技术借鉴于迟浩师兄的pFind,谱图预处理采用邬龙师兄专门训练的 pParse,15N标记检验需要刘超师兄的pQuant,FDR论证和部分质量控制方法借鉴于文锋师兄的pGlyco和文婧师姐的pValid。 pLink 2 也继承了pLink 1的部分算法和软件,特别是BS3和SS的两个标注集,这是当年吴妍洁师姐和樊盛博师兄的贡献。樊师兄读博士期间兢兢业业维护pLink 1软件,这才有了众多CNS应用文章,使我看到了pLink的价值,进而激励我也兢兢业业维护pLink 2软件。特别感谢贺老师,如果不是贺老师不断推着我前进,不断地对论文提出更高的要求,文章的质量肯定达不到现在的水平,审稿的过程也不会像现在这么顺利。
pLink 2的问世也离不开董老师实验室师生的长期合作。在pLink的研究与开发工作中,董老师实验室一直以来都是我们最密切的合作伙伴,杨兵师兄和卢珊师姐早年制备的合成肽段交联数据集支持了pLink 1和 pLink 2灵敏度优势的确立,丁曰和师兄、谭丹师姐和曹勇师兄为此次投稿制备的两批15N标记数据集支持了pLink 2准确度优势的论证。特别感谢当年董老师带领大家完成了pLink 1连续突破Nature Methods,现在我们照虎画猫完成pLink 2突破Nature Communications。
pLink 2的发表,不是休止符。正如论文Discussion中所写,交联质谱数据的深度解析和精准鉴定还远没有达到,pLink的速度和精度还有很大的提升空间。此外,面对越来越广泛使用的可碎裂交联剂, pLink需要及早支持。与pFind相比,pLink是一种更复杂的开放式修饰鉴定,Open-pFind所能达到的功能、速度和精度将会是未来pLink的努力目标。另外,目前pLink的应用成果主要源自 pLink 1软件,随着 pLink 2文章的发表,相信会有更多应用成果源自pLink 2软件,我将一如既往做好软件维护和技术支持,帮助更多的生物学家、化学家们完成他们的研究,发表更多的CNS文章:-)。
最后,总结一下我发表第一篇科研论文的经验与教训。回望整个研究历程,pLink 2的工作从2012年开始,前后历经三届学生,直到2019年才正式发表。无论对于pLink 2来说,还是对于参战师生来说,这个历程都不算短。漫长的科研道路充满坎坷,往往解决完一个问题后又出现另一个问题,这要求我们具备不屈不挠的精神,坚持不懈地与困难作斗争,借用毛主席的一句话就是“最后的胜利,往往在于再坚持一下的努力之中”。另一方面,我个人在 pLink 2的研究过程中,常常因为科研压力而焦虑沮丧,甚至有一段时间因为贺老师对论文的修改建议而失眠。现在,当我回想起这一路上的心情时,觉得大部分负面情绪是完全可以避免的,因为无论是导师的建议还是评审人的意见,大都是为了帮我们提升论文质量。如果以这种思维方式来看待我们遇到的困难与批评,科研之路也许会更加快乐与持久。
想起当年我刚接手pLink 2工作时,刘超师兄说过一句话:“pLink 2的相关工作,需要一个像艾森豪威尔那样细致工作的人,把林林总总、方方面面、繁繁琐琐的细节落实到位。”以上就是我当艾森豪威尔的经历:-)。
2019年9月30日