pFind 2.6 User Guide
Contents
3.3 Setting advanced parameters
1 Program description
pFind is a search engine for automated peptide and protein identification from tandem mass spectra. It can run on both MS Windows and Linux operating systems. The Windows version of pFind has a graphical user interface(GUI) and the command-line programs. The Linux version only has the command-line programs.
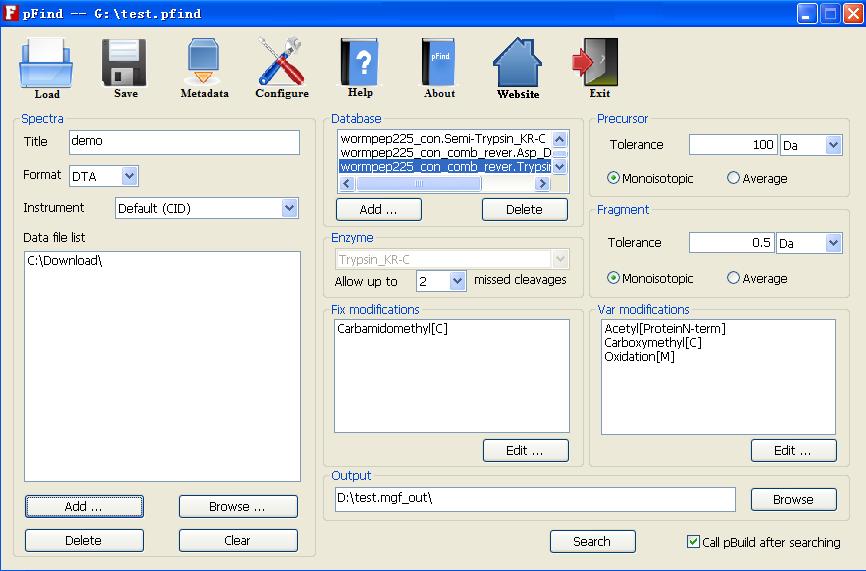
Fig. 1 The GUI of Windows version of pFind
2 Installation
2.1 Hardware requirements:
CPU: 1.0 GHz or higher
Memory: 1 GB or higher recommended
Hard Disk: 10 GB or higher
2.2 Installation on Windows
The Windows setup package of pFind Studio can be downloaded from the website http://pfind.ict.ac.cn. Before installation, please fill in a registered table and send it to pfind@ict.ac.cn to get a registration key.
The pFind Studio setup package includes not only pFind, but also pXtract, pBuild, pScan and pLabel. pXtract creates .DTA .MGF and .MS2 input files directly from Thermo Scientific .raw LC-MS/MS data files. pScan is a flexible tool that helps biologists to preprocess protein sequence databases in proteomics research. pBuild is a tool that can compare several search engines' results and combine them together. The latest version, pBuild v2.0, can process the search results of pFind, SEQUEST and Mascot. pLabel is a spectra labeling tool that can visualize the global- and local-view peptide-spectrum matches, given the results of pFind or any other search engines. pLabel can label both CID and ETD spectra, and implement the manual de novo sequencing.
2.3 Installation on Linux
Run the installation script first, than add the path of pFind installation into the environment variables of “PATH” and “LD_LIBARAY_PATH”. This can be accomplished by adding the follow lines into the existing .bashrc file in user default directory.
export PATH:=/usr/pfind/bin:&PATH
export LD_LIBARAY_PATH:=/usr/pfind/bin:&LD_LIBARAY_PATH
3 Usage
The general approach for search is to take a small sample of the protein of interest and digest it with a proteolytic enzyme, such as trypsin. The resulting digest mixture is analysed by mass spectrometry.
The mass spectrometer will measure a set of molecular weights for the intact mixture of peptides. Then the MS/MS can provide structural information by recording the fragment ion spectrum of a peptide. Usually, the digest mixture will be separated by chromatography prior to MS/MS analysis, so that MS/MS spectra from individual peptides can be measured.
The experimental mass values are then compared with calculated fragment ion mass values, obtained by applying cleavage rules to the sequences in a database. By using the scoring algorithm, the best match or matches can be identified.
The parameters and operations of pFind are detailed in the following sections.
3.1 Startup GUI
Double click the icon ![]() , the
pFind will start up. You will see a welcome screen. After that, the main dialog
window of pFind will display.
, the
pFind will start up. You will see a welcome screen. After that, the main dialog
window of pFind will display.

Fig. 2 The welcome screen of pFind
3.2 Setting common parameters
3.2.1 Spectra
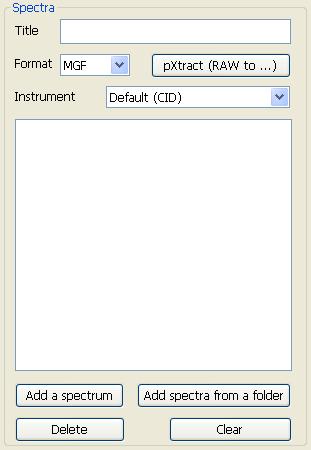
Fig. 3 The parameters for tandem mass spectra
Format
Following formats are supported by pFind:
l Micromass (.PKL)
l Sequest (.DTA and .DTAS)
l Mascot (.MGF)
l Yates Lab(.MS2)
l Finnigan (.RAW)
pXtract can be used to transform the tandem mass spectra from one format to another format.
Path
The path or folder containing the tandem mass spectra.
Instrument
Instrument determines which fragment ion series will be used for scoring. When “UNKNOWN” is selected, the default ion series (B+, B++, Y+, Y++) will be used. Users can view the configuration file “instrument.ini” in the installation folder of pFind to add, to lookup or to edit the fragment ion series setting for each instrument type.
3.2.2 Database and enzyme
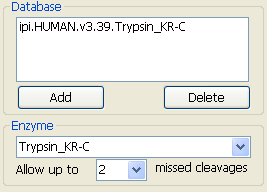
Fig. 4 The parameters about database and enzyme
Database
The sequence database to be searched.
Enzyme
The enzyme type and the maximal number of allowed missed cleavage sites for in vitro digestion of database sequences.
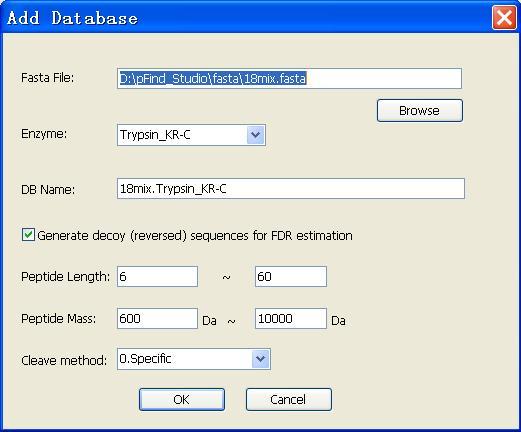
Fig. 5 The parameters about database adding
Add a new database
A new protein database (*.fasta) can be added by following steps:
l Click the “Add” button
l Select the path of the FASTA file
l Fill in the parameters of the database, and click the “OK” button
You can generate decoy (reversed) sequences for FDR estimation whose names begin with "REVERSE_".
3.2.3 Precursor and fragment

Fig. 6 The parameters about precursor and fragment
Precursor
These parameters are used to specify the error window tolerance of precursor peptide as well as the experimental mass value types, i.e., average or monoisotopic.
Fragment
The parameters used to specify the error window tolerance of precursor peptide. User also need specify whether the experimental mass values are average or monoisotopic.
The unit of tolerance
The mass tolerance is based on experimental precursor peptide mass values. Units can be selected from:
% fraction expressed as a percentage
mmu absolute milli-mass units, i.e. units of .001 Da
ppm fraction expressed as parts per million
Da absolute units of Da
3.2.4 Modifications
Most protein samples have modifications. Some modifications, e.g. phosphorylation and glycosylation, are post-translational modifications, while some modifications, e.g., oxidation, are artefacts of sample handling. Other modifications, such as cysteine derivatisation, are deliberately introduced during sample work-up.
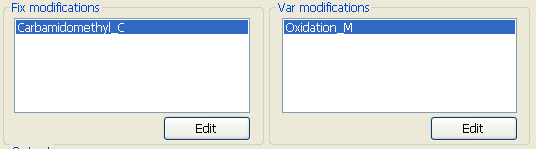
Fig. 7 The parameters about modifications
pFind supports two types of modifications. Fixed modifications are applied universally, to every instance of the specified residues or terminus. Variable modifications are those which may or may not be present. pFind enumerates all possible combinations of variable modifications to find the best match. Variable modifications can be a very powerful means of finding a match, but there are also dangers that sould be aware of. A search with many variable modifications can take a much longer time than the same search with fixed modifications.
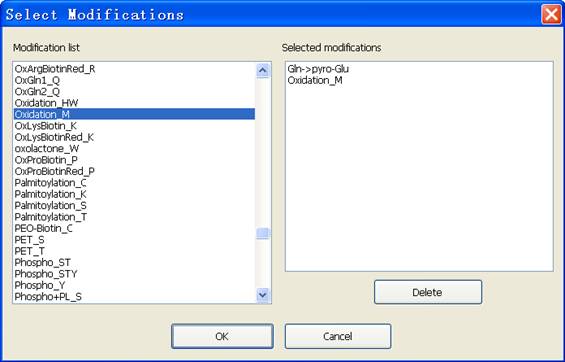
Fig. 8 The dialog of modification selection
3.2.5 Output

Fig. 9 The parameters about result output
Output specifies the folder of search results.
3.3 Setting advanced parameters
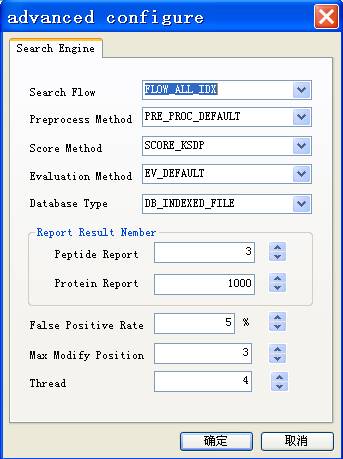
Fig. 10 The dialog of advanced parameters
Search flow
Specify the modes in which pFind workflow, default as FLOW_ALL_IDX.
Preprocessing method
Specify the preprocessing algorithms, with PRE_PROC_DEFAULT as default .
Scoring method
Specify the scoring algorithms, default as SCORE_KSDP. The experimental mass values are then compared with calculated fragment ion mass values, obtained by applying cleavage rules to the entries in a database. By using the scoring algorithm, the closest match or matches can be identified.
Evaluation method
Specify the evaluation algorithms, default as EV_DEFAULT.
Report result number
Specify the numbers of report peptides and proteins.
False discovery rate:
Specify the value of false positive rate, default as 1%. Users can input the value or use the scroll bar on the right to adjust this value. It CANNOT exceed 5%.
Max modify position:
Specify the maximal number of modification sites per peptide, default as 3.
Thread:
Specify the number of threads of pFind search engine for the multi-core CPU, default as 4.
3.4 Setting meta data
3.4.1 Modification
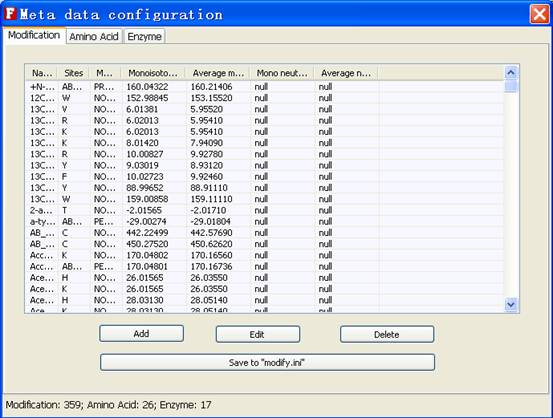
Fig. 11 The tab of modifications
In the modification tab, user can add, edit and delete the each entry of modifications, as shown in Figure 12.
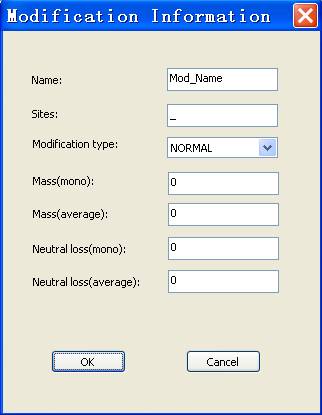
Fig. 12 The dialog of modification Information
3.4.2 Amino Acid
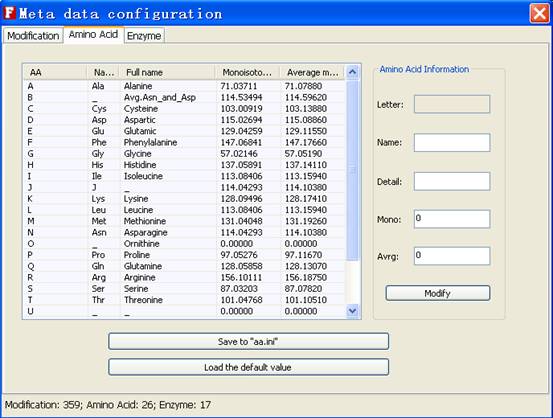
Fig. 13 The tab of amino acid
In the amino acid tab, user can add, edit and delete the each entry of amino acid.
3.4.3 Enzyme
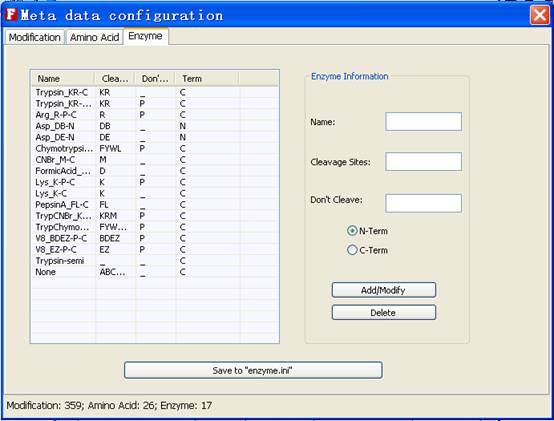
Fig. 14 The tab of amino acid
In the enzyme tab, users can add, edit and delete the each entry of enzymes.
3.5 Search and presentation
After a search is completed, a summary report is displayed that provides an overview of the results.
4 FAQ
l How can I obtain a copy of pFind?
You can download the setup package from our website: http://pfind.ict.ac.cn
l What platforms are supported by pFind?
pFind has been ported to Microsoft Windows 2000, XP, 2003, Vista, 2008, and 7, and Linux. In general, we support the current releases of these operating systems.
l How to analyze the search results of pFind?
You can use the pBuild, a tool that can compare several search engines' results and combine them together. pBuild can process not only the search results of pFind, but also the search results of Mascot and SEQUEST. pBuild also can be downloaded from our website.
5 Contact information
E-mail: pfind@ict.ac.cn
Address: Institute of Computing Technology, Chinese Academy of Sciences.
No. 6, Kexueyuan Sou th Road, Zhongguancun, Haidian District,
Beijing 100190, P.R.China
6 Publications
l Le-heng Wang, De-Quan Li, Yan Fu, Hai-Peng Wang, Jing-Fen Zhang, Zuo-Fei Yuan, Rui-Xiang Sun, Rong Zeng, Si-Min He and Wen Gao. pFind 2.0: a software package for peptide and protein identification via tandem mass spectrometry. Rapid Communications in Mass Spectrometry (RCMS) Vol.21, No.18, pp2985-2991, 2007.
l Dequan Li, Yan Fu, Ruixiang Sun, Charles X. Ling, Yonggang Wei, Hu Zhou, Rong Zeng, Qiang Yang, Simin He and Wen Gao. pFind: a novel database-searching software system for automated peptide and protein identification via tandem mass spectrometry. Bioinformatics, 21(13), 3049-3050, 2005
l Yan Fu, Qiang Yang, Ruixiang Sun, Dequan Li, Rong Zeng, Charles X. Ling, Wen Gao. Exploiting the kernel trick to correlate fragment ions for peptide identification via tandem mass spectrometry. Bioinformatics 20, 1948-1954, 2004