pFind Studio: a computational solution for mass spectrometry-based proteomics
Introduction
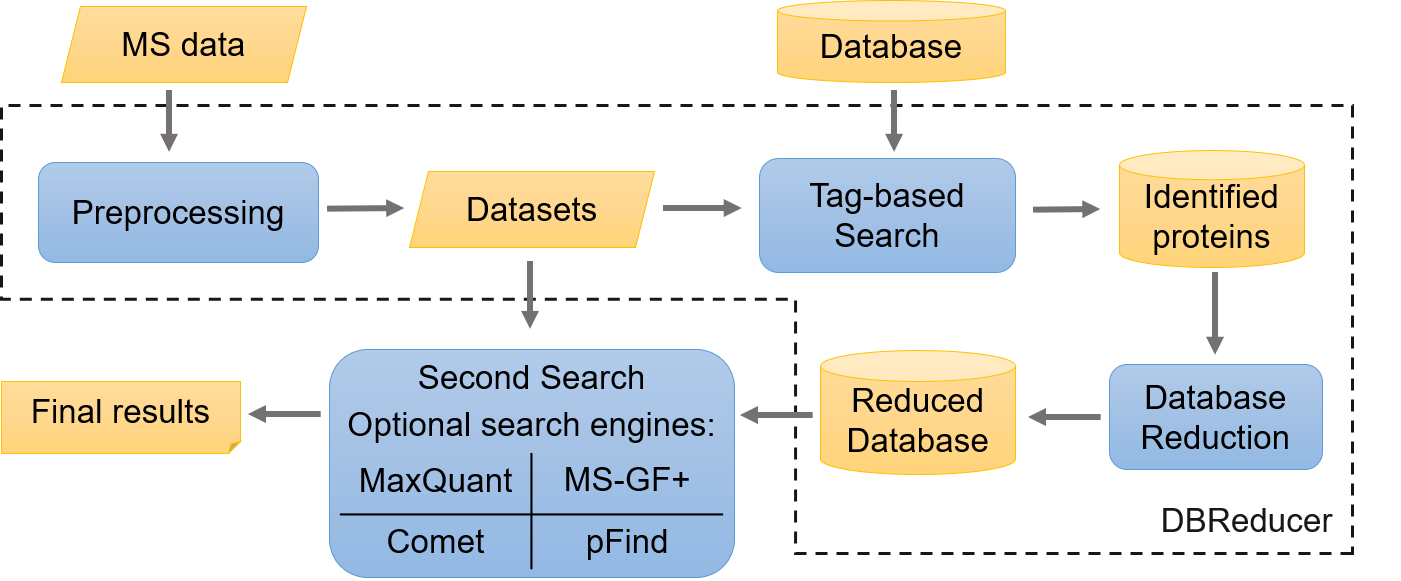
Mass spectrometry-based metaproteomic and proteogenomic studies tend to use large-scale databases which may contain too many irrelevant or suspect proteins, and such an imprecise database presents challenges for both the accuracy of peptide identifications and the time consumption. To address them, we developed a sequence tag-based strategy for database reduction, DBReducer, to efficiently reduce databases for iterative database search. DBReducer can be a database reduction method prior to common proteomic analysis, especially for scenarios with the large-scale database.
Downloads
DBReducer version 1.0 is currently free to use. click to download.
For source code and guideline, please refer to github.
For other issues, please contact wangkaifei20g@ict.ac.cn.